



细胞坏死——细胞死亡研究新热点
时间:2020-11-18 10:35:02 浏览次数:298



19年小优写了一篇细胞焦亡的文章(感兴趣的小伙伴点这里),大家看完之后有没有感觉涨芝士啦。
Whatever,小优和大家每天进步一点点,SCI实现不是梦。今天给大家带来了细胞死亡之坏死的分享呀。
往下点有彩蛋哦


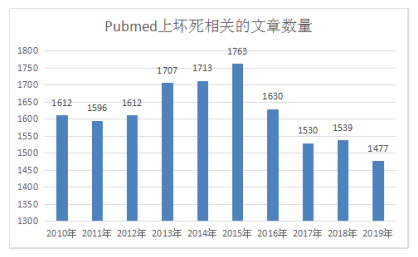
小优也整理了下Pubmed上近10年来坏死相关的文章。
从图中可以看出来,坏死虽然近几年来热点在下降,但是也有可能逆袭成为下一个研究的风向标哦,毕竟每年CNS上依旧很多坏死相关的高分文章。

突突突突突突......来不及解释了快上车
在详细了解坏死之前,小优先带大家复习下呀。
细胞死亡分为凋亡(Apoptosis),坏死(Necroptosis),焦亡(Pyroptosis),自噬性死亡(Autophagic cell death)等死亡方式。
那接下来小优考一下大家啦,大家知道以下一组图片分别对应的是细胞的哪种死亡方式吗?
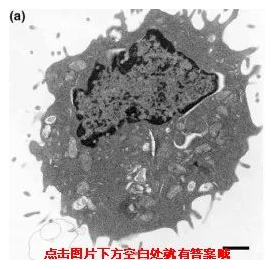
正常
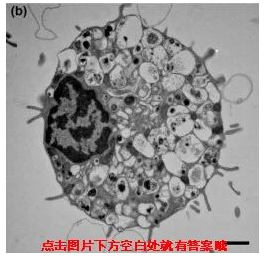
自噬
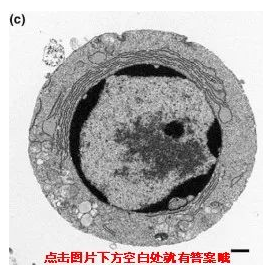
凋亡
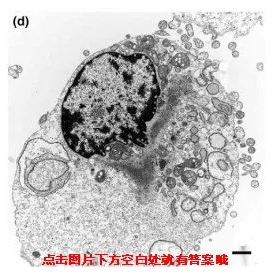
坏死
凋亡是生理条件下的基因调控,自噬主要是营养缺乏或者激素诱导,坏死主要是由极端的物理化学因素或者严重的病理性刺激诱发,除此之外,凋亡、焦亡等也会引起坏死。
不同死亡方式,电镜下呈现出来的细胞形态也是不同的,发生凋亡的细胞会变小,自噬的细胞产生空泡,坏死细胞膨大、变形,除此之外凋亡和自噬的细胞膜是完整,坏死细胞的细胞膜结果被破坏。
各位小伙伴们知道了以上图片对应的死亡方式了吗?答完题接下来,进入我们今天的重点啦!小优特意整理了出了几篇关于细胞坏死的热门文章跟大家分享一下。

01
肠道干细胞基因组不稳定引发干细胞坏死,导致自发性肠炎
在真核生物的基因组中,存在一系列叫做内源性逆转录病毒(ERVs)的不编码序列,它在人体内含量也很丰富。ERVs虽然不编码,但它的活跃会引起基因组不稳定以及激活细胞内固有免疫。绝大多数ERVs需要被DNA甲基化或组蛋白甲基化修饰,使其处于静默状态,从而维持细胞的稳态。
这篇文章发现在健康的肠干细胞中,SETDB1(H3K9位点三甲基化甲基化转移酶)对ERV区域进行特定的组蛋白三甲基化修饰,从而抑制ERV的激活,保证基因组的稳定性,维持细胞的稳态,保证机体的健康。而在炎性肠炎(Inflammatory bowel disease,IBD)病人的样品中,在肠干细胞区域SETDB1表达量明显下降,与此同时,在小鼠肠道上皮或者仅在肠道干细胞中敲出Setdb1后,也会产生类似于人IBD的自发性炎症。本文通过敲除Setdb1引起小鼠肠道炎症,引起的基因组不稳定性和后期肠道炎症驱动干细胞死亡。
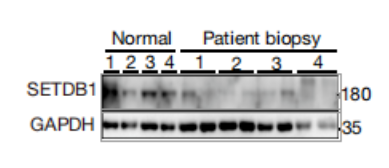
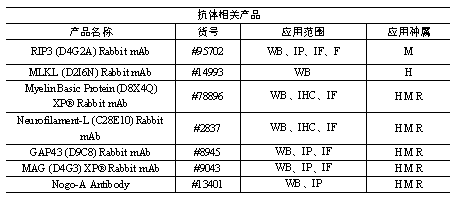
正常肠组织(N0rmal)和IBD患者的活检(Patient biopsy)中的SETDB1蛋白WB结果。具有相同患者编号的两条泳道是在内窥镜下同时采集的两次活检。
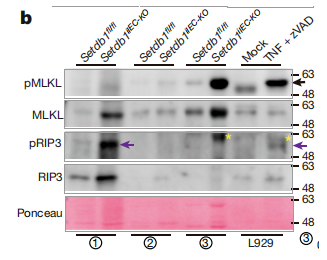
Western blot在Setdb1敲除的隐窝中检测到了磷酸化的RIP3和磷酸化的MLKL(坏死的标志)
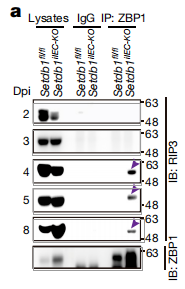
在样品的IP实验显示,ZBP1与RIP3相互作用激活坏死
在鉴定细胞死亡类型的研究中,作者发现只有在肠炎小鼠中将necroptosis通路中的RIP3或MLKL敲除,可以阻止肠道干细胞的死亡。经典坏死通路的信号来自于细胞外肿瘤坏死因子(TNF)并依赖于RIP1的激酶活性。在坏死性凋亡中,RIP1和RIP3,RIP3和RIP3间通过两者共有的RHIM结构域相互结合,而ZBP1是另外一个具有RHIM结构域的蛋白。之前的研究发现一些病毒,比如流感病毒IAV,小鼠巨细胞病毒(MCMV)可以通过ZBP1激活坏死。在非感染情况下,ZBP1介导的坏死只有在RIP1的RHIM结构域突变的小鼠上发现【11】,但是其生物学意义和机制仍是未知。
本文中作者通过对转录组的KEGG分析,发现在Setdb1敲除的肠干细胞中,有大量类病毒的积累。通过对结合在ZBP1上的核酸进行测序分析,发现这些核酸是ERV RNA。ZBP1的N端有结合核酸的结构域,如果将此结构域剔除,Setdb1敲除导致的肠干细胞死亡可被抑制。由此认为,Setdb1的缺失使得ERV重新活跃,破坏了基因组的稳定性,大量积累的ERV RNA作为死亡信号,结合并激活ZBP1启动坏死性凋亡。这样的结果证明了ZBP1介导的坏死在非感染性炎症中重要病理作用。
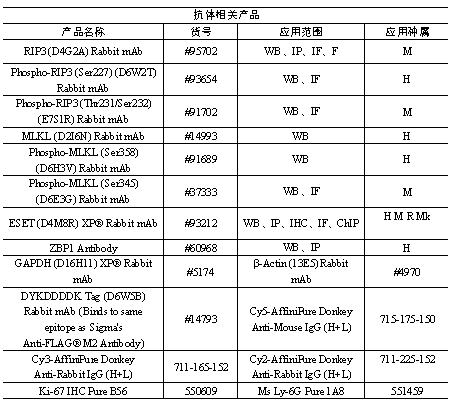
小优也帮大家整理出来本篇文章中用的产品哦,如果您也正在做或者即将做进一步的研究,需要相关的产品,可以随时联系小优哦!
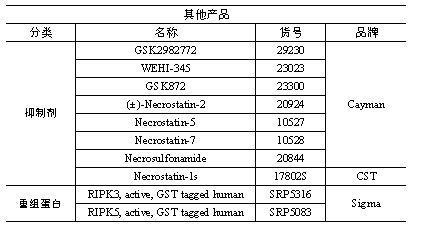
02
神经髓鞘降解机制--细胞坏死的关键蛋白MLKL的新功能
髓鞘(myelin)是包绕在神经元轴突外部的多层膜组织,在中枢神经系统中由少突胶质细胞产生,在外周神经系统中由施旺氏细胞(Schwann cell)产生。它的主要功能是保护轴突、通过绝缘作用使动作电位在轴突的传导加快、在神经损伤后调节轴突的再生等。髓鞘的降解发生在脱髓鞘性疾病和神经损伤中。在脱髓鞘性疾病中,髓鞘由于遗传、感染或自身免疫等原因异常丢失,造成轴突传递动作电位的效率下降和轴突退变。在神经损伤后,需要及时清除髓鞘,以促进轴突再生。
本篇文章中,作者同时运用Rip3和Mlkl基因敲除小鼠研究细胞坏死在机体内的生理学意义。研究发现细胞坏死信号通路调控雄性小鼠生殖系统衰老,如果使用RIP1抑制剂则可以阻止雄性小鼠生殖系统的衰老。除了阻止衰老之外,那么围绕RIP1/RIP3/MLKL通路的这些关键蛋白是否还有新的重要的未知生物学功能呢?
在这项研究中发现MLKL在坐骨神经损伤后执行髓鞘的降解,Mlkl敲除小鼠的坐骨神经髓鞘降解速率显著低于野生型小鼠。MLKL尽管作为RIP3的底物,但是Mlkl敲除小鼠的表型和Rip3敲除的小鼠在介导髓鞘降解上的表型并不相同,Rip3敲除的小鼠并没有表现出髓鞘降解的表型,而且使用RIP1的抑制剂也不起作用,也就是说MLKL在髓鞘降解上功能不依赖于上游激酶RIP3的激活。在神经损伤信号存在的情况下,MLKL在施旺氏细胞中表达并通过第441位丝氨酸磷酸化激活,继而通过对脑苷脂的结合嵌入到髓鞘的膜结构中,从而降解髓鞘。文章同时证明了MLKL介导的髓鞘降解对于神经轴突的再生是必需的。
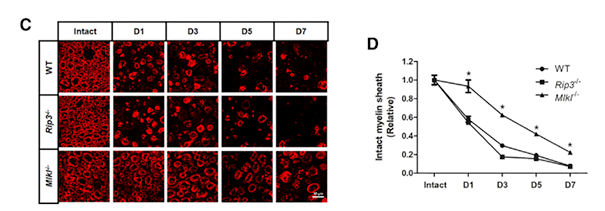
分别对野生型、Rip敲除型和Mlkl敲除型损伤的神经横断面MBP进行免疫染色,发现MLKL的表达显著降低髓鞘的降解速率
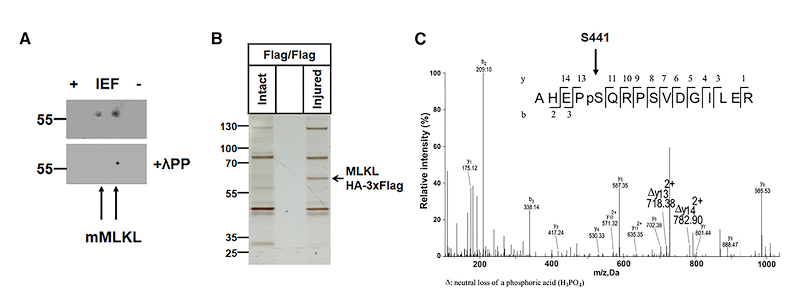
通过对损伤神经进行2D电泳,以及对MLKL HA-3XFlagde 免疫沉淀和质朴分析,结果发现MLKL在丝氨酸441位发生磷酸化。
总的来说,该研究发现了细胞坏死关键蛋白MLKL非依赖RIP3激活的新功能,为神经损伤后的恢复及脱髓鞘疾病的治疗提供了新的思路。
03
细胞凋亡,坏死和自噬等主要细胞死亡形式的分析

癌症一直是世纪难题,为了可以更有针对性的开发细胞异常死亡导致的癌症等相关疾病的有效治疗方法,必须把各种细胞死亡方式研究清楚。本文作者详细的总结阐述了凋亡、焦亡以及坏死等各种不同细胞死亡方式和细胞特征等,加深广大科研工作者对细胞死亡各种形式的认识,未来可以开发出针对癌症等疾病更有效的临床疗法。
细胞凋亡是为维持内环境稳定,在一定时间内,细胞按一定的程序发生死亡,这种细胞死亡具有严格的基因时空性和选择性。凋亡缺陷会导致肿瘤等细胞死亡障碍疾病的发生,在某些情况下如心脏受袭也会导致凋亡过激从而杀死许多无辜细胞。
细胞凋亡涉及一系列基因的激活、表达以及调控等,其特征在于质膜不对称性和附着的丧失,核质浓缩,核膜、核仁碎裂,DNA 片段化和 mRNA 衰变等,早期特征之一是质膜的改变。
凋亡可被多种细胞信号激活,如钙稳态失衡、氧化损伤、线粒体损伤、毒素、生长因素和激素刺激等。细胞凋亡过程大致分为:凋亡信号转导→凋亡基因激活→细胞凋亡的执行→凋亡细胞的清除。启动细胞凋亡的机制可分为内源性信号通路和外源性信号通路两种,即线粒体途径和死亡受体途径,这两种途径分别由Bcl-2 蛋白家族和细胞表面的“死亡受体”与其对应的配体所调控。
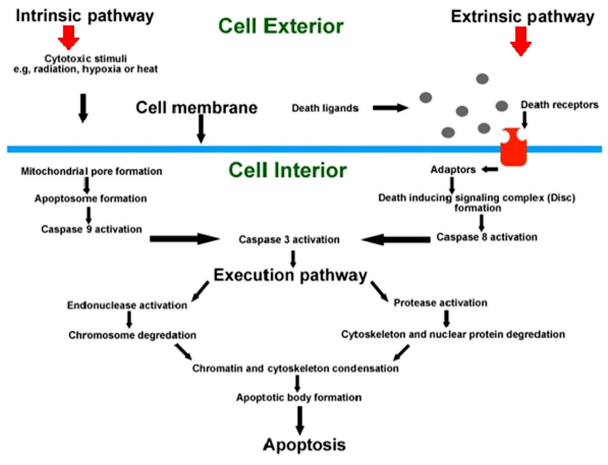
细胞凋亡的内源性途径是由细胞自身响应损伤而引发的。外源性途径是通过免疫系统细胞刺激的死亡受体启动的。当激活半胱天冬酶3,两种途径都汇合,从而导致细胞凋亡。
与凋亡不同,坏死是由外部因子(如缺氧或炎症)引起的另一种不受控制的细胞死亡形式。此过程通常涉及各种促炎性蛋白和物质(例如核因子κB)的上调,细胞膜破裂等特征。与细胞凋亡相反,坏死是细胞死亡的一种与能量无关的形式,细胞被突然的刺激(辐射,热,化学物质,缺氧等)严重破坏,无法正常运作。坏死细胞的膜通透性增高,致使细胞肿胀,细胞器变形或肿大,坏死早期细胞核无明显形态学变化,但是最后细胞破裂,细胞裂解释放出内含物,引起炎症反应和组织损伤。
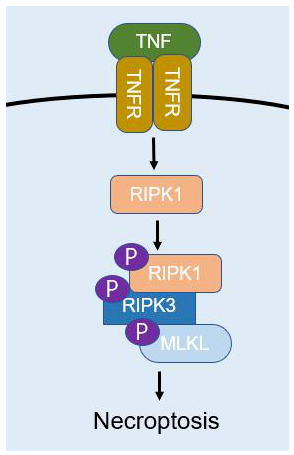
之前研究就发现了调控坏死的关键因子RIPK1、RIPK3 和 MLKL。RIPK1 的激酶活性对于多种复合体至关重要,这些复合体能够调节炎症(复合体 I)、凋亡(复合体 IIa)和坏死性凋亡(复合体 IIb)。RIPK1 泛素化导致 NF-κB 激活及后续的炎症级联,以及 RIPK1 在 Ser320 位点的磷酸化。去泛素化酶(如 CYLD 和 A20)能够抑制复合体 I,从而促进 RIPK1 与 caspase-8 以及诱导死亡的复合体 IIa 通路的相互作用。与 RIPK3 的相互作用为复合体 IIb 信号转导和坏死性凋亡所必需。Caspase-8 活性可通过剪切 RIPK1 和 RIPK3 来抑制坏死性凋亡。caspase-8 活性的抑制能够防止 RIPK1的剪切并驱动坏死性凋亡。在坏死性凋亡期间,RIPK3在 Ser227位点被磷酸化,此磷酸化为MLKL的激活所必需,MLKL作为 RIPK1 和 RIPK3 下游的效应蛋白发挥作用。这些活动是复合体 IIb 的一部分,也称为坏死小体。MLKL磷酸化会诱导 MLKL 寡聚化并向质膜转位,并与磷脂酰肌醇相互作用,诱导膜通透和细胞破坏。MLKL诱导的质膜形成孔道导致通透性增高,释放细胞损伤相关分子如线粒体 DNA和白介素 (IL)-33等。

坏死性凋亡信号通路涉及TNF信号通路和RIP1的去泛素化,RIP1和RIP3的磷酸化,caspase 8的失活以及MLKL的磷酸化,最终引起坏死性凋亡。
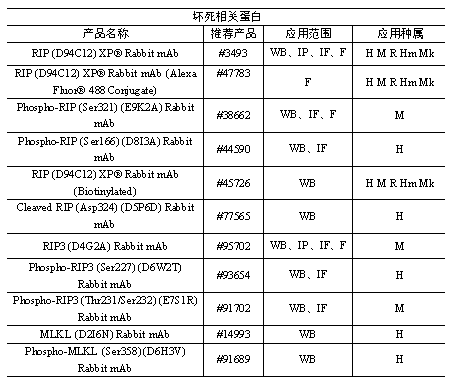
本篇文章中涉及到的产品全是信号通路相关的哦,如果您正在或者即将要做坏死的话,作为细胞信号通路研究金标准的CST是您的不二之选哦。
小优给大家列出了部分坏死相关的产品清单哦。
上海优宁维生物科技股份有限公司
试剂 | 耗材 | 仪器 | 软件 | 定制 | 实验服务 | 供应链
免费热线:4008-168-068
咨询邮箱:info@univ-bio.com
订购商城:www.univ-bio.com
微信公众平台:优宁维抗体专家,欢迎关注!
小优博士(小程序):5大课堂, 让你的科研不再难!



 危险品化学品经营许可证(不带存储) 许可证编号:沪(杨)应急管危经许[2022]202944(QY)
危险品化学品经营许可证(不带存储) 许可证编号:沪(杨)应急管危经许[2022]202944(QY)  营业执照(三证合一)
营业执照(三证合一)