





炎症小体在2002年被发现以后就一直是免疫和炎症疾病领域的重点研究对象,研究热度逐年攀升。

那么炎症小体是什么呢,为什么大家都热衷于研究它呢?
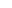
炎性小体是一种蛋白质复合物,主要包含三部分,受体蛋白、接头蛋白ASC以及下游的Caspase家族。受体蛋白分为NOD样受体(NLR)家族和PYHIN家族。NLR家族大家会比较熟悉,包括NLRP1,NLRP2,NLRP3,NLRP6,NLRC4以及NLRP12。它们的共同点是都包含核苷酸结合结构域(NBD),富含亮氨酸的重复序列(LRR)。有的NLR还包含热蛋白结构域(PYD)或caspase激活和募集结构域(CARD)或者全都要。

PYHIN家族成员相对来说研究少一些,AIM2和IFI16的特征在于,除了PYD之外,还具有HIN200结构域,参与配体结合。不过研究的少才能突出创新性,今年AIM2炎症小体也发了两篇Nature,人少好办事嘛。

接头蛋白ASC主要有两个结构域,一个PYD,一个CARD,ASC在这里起到桥梁作用,连接受体蛋白和效应蛋白caspase。Caspase含有CARD结构域会被招募激活,但并不是所有的caspase都会被招募,主要是caspase-1,4,5,11,12参与促炎作用。

图1 炎症小体的结构
那么炎症小体的功能是什么呢?
前面我们讲到炎症小体可以招募pro-caspase-1,调控caspase-1的活性并激活caspase-1。caspase-1被激活后,会将无活性的促炎细胞因子pro-IL-1β、pro-IL-18切割为成熟体IL-1β和IL-18。炎症小体就是个平台,加工IL-1β和IL-18,并介导细胞焦亡。你问我细胞焦亡是什么?
细胞焦亡是一种细胞程序性死亡,其表现特征与细胞凋亡、细胞坏死有部分重叠,比如DNA损伤、TUNEL染色阳性、Annexin V染色阳性、细胞膜上孔隙形成、细胞裂解等,伴随着大量炎性细胞因子的释放。
那么炎症小体是如何形成呢?
不同的内源和外源信号可能会诱导组装不同的炎症小体。比如AIM2是一类DNA感受器,可以通过它C端HIN200结构域识别并结合自体或异体的DNA。再比如NLRC4炎性小体仅由特定的PAMP激活,而NLRP3炎性小体比较“包容”,它的配体就比较多样化:
a) 细胞外ATP诱导K+通过P2X7受体外排;
b) PAMPs和DAMPs触发活性氧(ROS) 的生成;
c) 细胞内形成的结晶或颗粒结构导致溶酶体破裂以及溶酶体内容物如组织蛋白酶B的释放;
d) 内质网应激(endoplasmic reticulum stress,ER stress) ;
e) 自噬功能障碍;
f) Ca2 +超载等。

图2 不同炎症小体的激活方式
那么炎症小体是好是坏呢?
有同学问,炎症小体是好是坏呢?我在实验中应该抑制还是激活呢?这里,小优想送大家一句话,“抛开剂量谈毒性都是耍流氓”——过犹不及。炎症小体的活动是需要严格控制的,不能随意抑制和刺激,以避免产生过多的细胞因子导致细胞死亡,伤及自身。所以正常情况下,炎症小体,特别是NLRP3的表达在许多细胞中相对较低,需要诱导去引发信号。这也解释了很多同学的疑问,为什么自己正常细胞/组织中检测不到NLRP3。
机体调节机制有如下几种:
1. 炎症小体蛋白会发生可变剪接,产生不同活性。比如ASC的其中一个剪接变体是具有抑制活性的。
2. 机体会表达一些调节炎症小体活性的蛋白质,主要是通过CARD或PYD的相互作用来分隔炎症小体成分组装或直接抑制caspase-1功能。比如ASC主要表达在静息细胞的细胞核中。
3. 炎症小体活性的调节也可以通过与细胞应激等(例如自噬)相互作用来实现。有研究发现,自噬缺陷细胞的炎症小体活化阈值降低了,可能是因为线粒体清除能力受损导致ROS水平升高。
4. 炎症小体活性调节的还可能通过分泌因子或细胞间相互作用而下调。例如I型干扰素或CD4+ T细胞与巨噬细胞或树突状细胞之间的相互作用,都导致炎症小体活性下调。
总之,我们的机体已经进化出不同的机制来调节炎性体的活化,以防止炎性体的过度活化。
在各种疾病中,炎性小体究竟发挥了什么作用呢?
微生物
研究最广泛的NLRP3炎症小体,已经被证明与抗菌,病毒,真菌和寄生虫的免疫反应有关,不过大部分情况是间接识别的。许多研究表明微生物是通过诱导与细胞应激和损伤有关的信号来激活炎症小体。例如甲型流感病毒,感染时借助Toll样受体7(TLR7)识别病毒RNA,从而诱导NLRP3炎症小体成分的转录。
在微生物感染期间,不同炎症小体的协同活性对于机体产生保护性免疫应答的能力非常重要。为了避免炎症小体激活,许多病原体进化出新的功能——编码炎症小体抑制剂。例如流感病毒的NS1或牛痘病毒的Crm1,这些抑制剂会作用于炎症小体的核心成分,并直接抑制caspase-1的活性。其他抑制剂,例如人类疱疹病毒的ORF63,可结合NLR并阻止NLRP1和NLRP3炎性小体的形成。这些微生物的免疫逃逸机制反而体现了炎症小体的重要性。接下来,我们再具体研究下炎症小体是如何感测病原体的,以及炎症小体如何调节免疫,我们就能在抵抗微生物的路上走宽了。
晶体沉积疾病
晶体沉积疾病是一种通过暴露于外源或内源性结晶分子引起的炎症性损伤。外源性分子,例如二氧化硅,它是通过肺巨噬细胞的内吞作用,从而导致NLRP3炎症小体活化,包括ROS增加和溶酶体失稳,导致矽肺。内源性分子,例如尿酸钠(MSU)异常形成的晶体,会导致巨噬细胞中的NLRP3炎症小体活化。痛风就有尿酸钠的积累,并且已经在临床试验中已经开始应用IL-1β阻断剂。在骨关节炎患者中,也有研究报道滑液中的尿酸水平与IL-1β和IL-18水平以及疾病严重程度呈正相关。不过这些都需要进行进一步的研究和临床试验,以确定炎症小体是否是有效的治疗靶点。
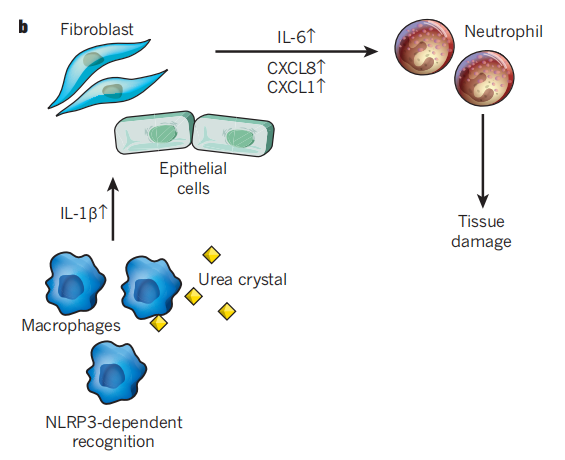
图3 尿酸晶体积累导致组织损失
胰岛素抵抗
在肥胖小鼠和人类的脂肪组织和肝脏中,NLRP3炎症小体成分和caspase-1激活增加。它们的表达水平直接与Ⅱ型糖尿病(T2DM)的严重程度相关。过度饮食除了使脂肪细胞肥大,脂肪组织也被活化的M1巨噬细胞浸润。NLRP3,ASC和caspase-1在脂肪组织浸润的巨噬细胞中优先表达,然后NLRP3炎症小体活化。

图4 炎症小体在脂肪、胰腺、动脉中的参与作用
炎症小体和胰岛素抵抗有什么关系呢?研究报道,IL-1β会通过胰岛素受体底物的直接丝氨酸磷酸化抑制胰岛素信号传导并诱导肿瘤坏死因子TNF-α(一种特征明确的胰岛素抵抗促进细胞因子)的表达,从而产生胰岛素抵抗。胰岛素抵抗的坏处我们应该不用多提了。据报道,Nlrp3-/-、Asc-/-和Casp1-/-小鼠体重和脂肪增加比野生小鼠减少,胰岛素抵抗也有所改善。
外周胰岛素抵抗导致的慢性高血糖症可以通过刺激胰腺β细胞分泌的胰岛素来补偿。但随着时间发展,局部炎症与葡萄糖的毒性作用会一起加速β细胞的衰老,使胰岛素分泌减少,胰岛素抵抗便发展为明显的Ⅱ型糖尿病。IL-1β优先在胰腺浸润性巨噬细胞表达,在长期暴露于高浓度葡萄糖的情况下,它成了β细胞死亡的关键驱动因素。IL-1受体拮抗剂(IL -1RA)是否可以改善β细胞功能,以及具体如何改善的,大家可以行动起来了。
动脉粥样硬化
动脉粥样硬化的炎性性质已得到充分证实,但是由什么因子来触发的,研究的还不够透彻。胆固醇的沉积是动脉粥样硬化的病理特征,最近的证据表明它们可能是通过弱氧化低密度脂蛋白(mmLDL)启动巨噬细胞的吞噬溶酶体失活来激活NLRP3炎症小体。有研究报道,NLRP3,ASC或IL-1α/β敲除的LDL受体缺陷小鼠(易患动脉粥样硬化)对动脉粥样硬化具有明显的抵抗力。动脉粥样硬化的另一种模型是载脂蛋白E(Apoe)-/-小鼠,喂食高脂饮食时会发展为严重的高胆固醇血症和自发性动脉粥样硬化。研究表明,IL-1受体缺陷型Apoe -/-小鼠和IL-1RA治疗的Apoe-/-小鼠表现出动脉粥样硬化减少,提示炎症小体在动脉粥样硬化中的重要作用。
炎症性肠病
在胃肠道粘膜中,仅单层上皮细胞将机体与巨大的微生物生态系统隔开。为了避免对共生微生物和食物抗原的持续炎症反应,同时保持对病原体侵害的反应能力,哺乳动物已经进化出由上皮和基质细胞以及造血细胞组成的复杂的粘膜免疫系统。这些细胞相互作用,合作密切。当这种相互作用受到干扰破坏时,可能会发生自发炎症,从而可能导致炎症性肠病(IBD)。
有的课题组发现炎症小体在在化学诱导的肠道自发性炎症中的重要参与作用,而有的课题组发现缺乏NLRP3,ASC和caspase-1的小鼠表现出病情加重,似乎前后矛盾。还有报道说,NLRP6炎性小体参与了共生菌群的稳态调节——肠道菌群的量子力学。还有人提出,炎症小体参与了炎症诱导的肿瘤发生;而另一篇报道表明上皮NLRC4炎性小体活性也可能参与了肠道炎症引起的肿瘤发生的预防,因为NLRC4缺陷型小鼠在诱发慢性炎症后更容易发生结肠肿瘤。其实我们简单理解一下就是要有话事人维持秩序,不然就乱了套。不过炎症小体的确切分子机制仍有待深入研究。
炎症小体虽然已经发现19年了,但是许多问题仍未得到解答,尤其是识别激活信号的机制。无尽的宝藏有待大家去深挖,帮忙动动手转给做炎症小体的同学们吧!
参考文献:
1. Till, et al. Inflammasomes in health and disease. Nature, 481(7381): 278-86.
2. Schroder, et al. The inflammasomes. Cell, 140, 821–832.
3. Davis, et al. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 29, 707–735.
4. Dinarello. IL-1: discoveries, controversies and future directions. Eur. J. Immunol. 40, 599–606.
5. Keller, et al. Active caspase-1 is a regulator of unconventional protein secretion. Cell 132, 818–831.
6. Bergsbaken, et al. Pyroptosis: host cell death and inflammation. Nature Rev. Microbiol. 7, 99–109.
7. Fernandes-Alnemri, et al. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature, 458, 509–513.
8. Hornung, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518.
9. Roberts, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323, 1057–1060.
10.Kerur, et al. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi sarcoma-associated herpesvirus infection. Cell Host Microbe 9, 363–375.
11.Zhou, et al. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nature Immunol. 11,136-140.
12.Hornung, et al. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur. J. Immunol. 40, 620–623.
13.Bryan, et al. Differential splicing of the apoptosis-associated speck like protein containing a caspase recruitment domain (ASC) regulates inflammasomes. J. Inflamm. 7, 23.

