






简介
哺乳动物表达系统的突出优点是表达蛋白可正确折叠以及进行糖基化等翻译后修饰,表达产物具有生物活性,已成为表达和生产蛋白质药物,基因工程抗体,疫苗抗原和研究用目的蛋白功能的首选表达体系。
哺乳动物表达体系蛋白表达的主要流程为:
PCR扩增目的片段——将基因片段克隆到表达载体上——细胞转染——细胞株的筛选和培养——纯化目的蛋白
解决方案
1、查找目的基因序列
由于引物是根据目的基因序列来设计的,所以查找到目的基因序列是设计引物的第一步。查找目的基因序列可以在多个网站上查到,也可以在文献中查找。
这里介绍的是比较常用的NCBI网站查找基因序列的方法。NCBI是美国国家生物技术信息中心,网站上具有多个生物数据库,是生物学研究常用的网站。
1.1打开NCBI网站,在下拉框选择Gene,输入目的蛋白名称;(这里以p65为例)
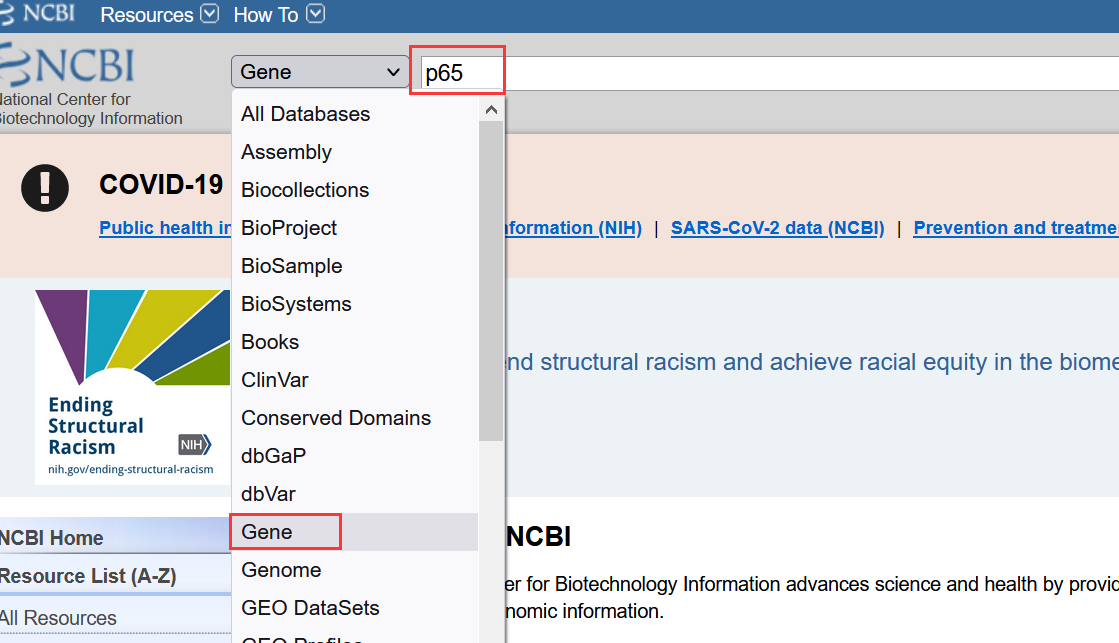
1.2 找到对应物种,也可以选择右侧框中的物种查找;

1.3 页面往下拉,找到mRNA and Protein(s),选择正确的isoform,NMxxx表示mRNA序列,NPxxx表示蛋白质序列;

1.4 新页面往下拉,点击CDS,点击右侧FASTA,即可得到目的基因的mRNA序列,复制粘贴到新的文本,以备使用。

2、选择合适的表达载体
常用的真核表达载体有pCMV,pcDNA等。表达载体一般包括启动子,多克隆位点,抗性基因,标签基因等等。
表达载体的选择原则:
1)启动子是否为真核启动子;
2)标签基因是否是我们需要用的标签。
常用的标签的标签有His,GST,MBP,Strep,Flag标签等,标签选择如下:

3、设计引物
设计引物也有多个软件可以使用,这里介绍的是常用的Primer 5软件。
设计引物的基本原则是:
1)引物长度一般为15-30bp,常用的为18-27bp,但不能大于38bp;
2)引物GC含量一般为40%-60%,以45-55%为宜,上下游引物GC含量和Tm值要保持接近;
3)引物所对应的模板序列的Tm值最好在72℃左右;
4)3'端最好不要是连续碱基,GGG或CCC会导致错误的引发,同时3'端最后一个碱基最好不要是A或T,否则容易导致错配;
5)以公式Tm=4*(G+C)+2*(A+T)-5计算Tm值,也就是退火温度。选择较低Tm值的引物的退火温度为反应的退火温度,最好保证每个引物的Tm值相匹配,且在70-75℃范围内;
6)在DNA测序和PCR中最好用5'末端稳定(GC含量多),而3'端不稳定(AT含量多)的引物,这种引物的结构可以有效地消除假引发反应;
7)引物和产物之间的Tm值相差别太大,20摄氏度范围内最好。
此外,在引物的两端需要加上酶切位点和保护碱基。为了保证基因序列的方向性,并且防止自连,现在基本上都选择双酶切位点。酶切位点的选择原则:
1)酶切位点存在于载体上,不存在于基因序列中;
2)两个酶最好有共同的缓冲体系;
3)两个酶在载体上酶切位点位置不能太近。
如果载体上没有标签序列,还需要在引物设计时加上标签序列。
如何确定酶切位点
3.1 打开Primer 5,选择file - new -DNAsequence,输入上述保存的CDS序列,点击As Is;
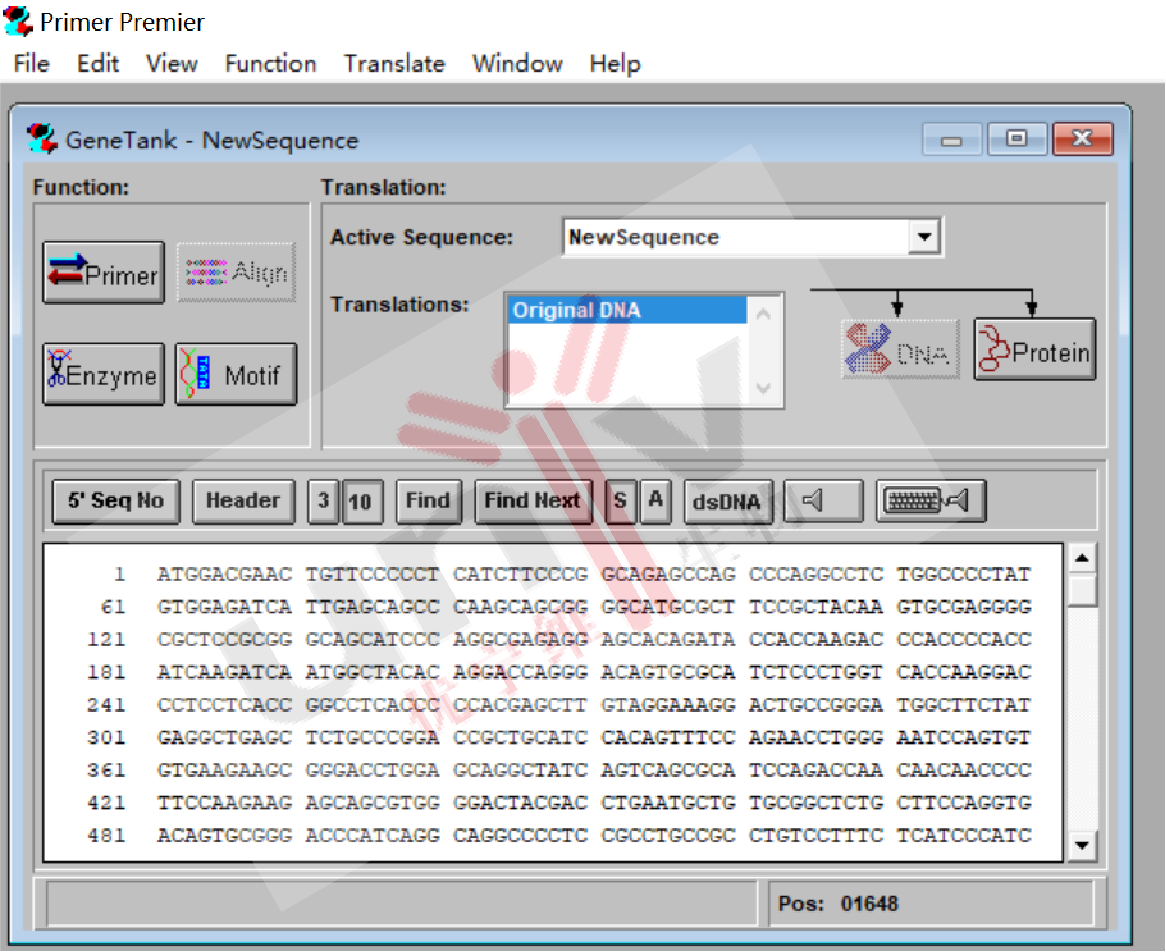
3.2 点击左侧Enzyme框,看一下载体上选择的双酶切位点是否在右侧框中,如果不在,从左侧框中添加到右侧框中;

3.3 点击OK即可查看CDS序列中有无选定的酶切位点,如果选定的双酶切位点存在于目的蛋白的CDS序列中,需重新选择双酶切位点。

如何设计引物
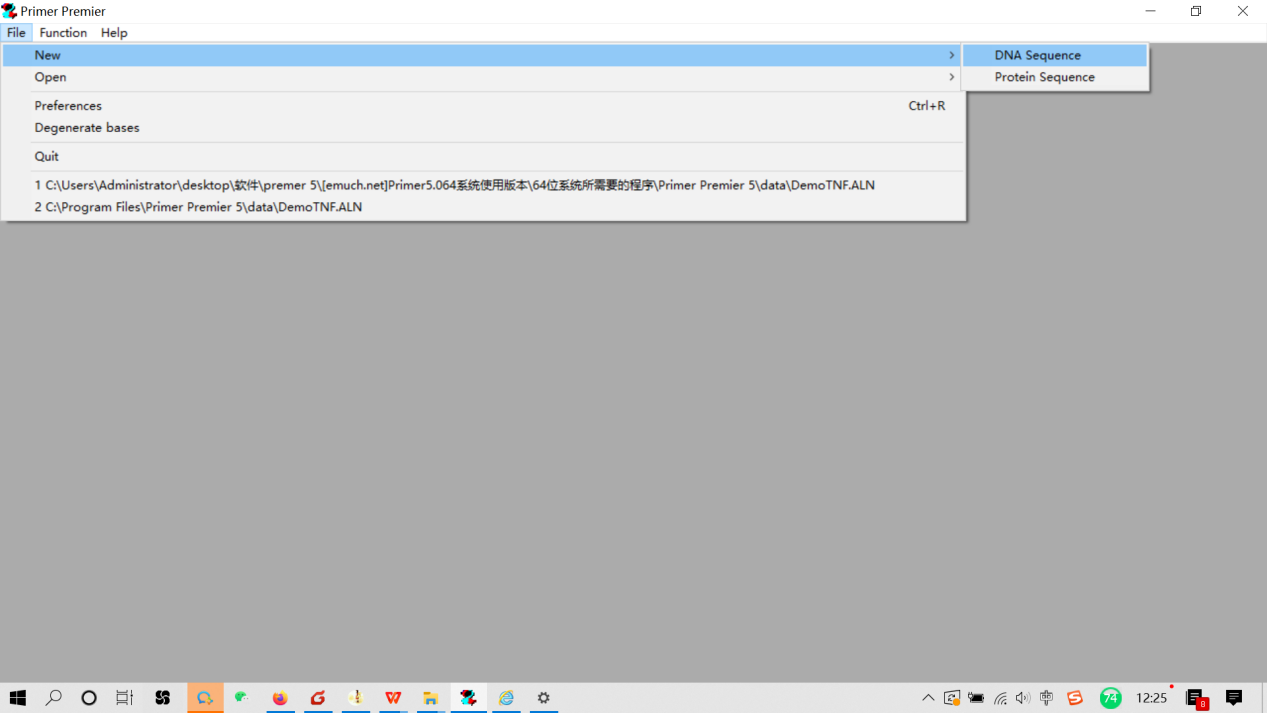
3.4 打开Prime 5软件,点击File——New——DNA Sequence
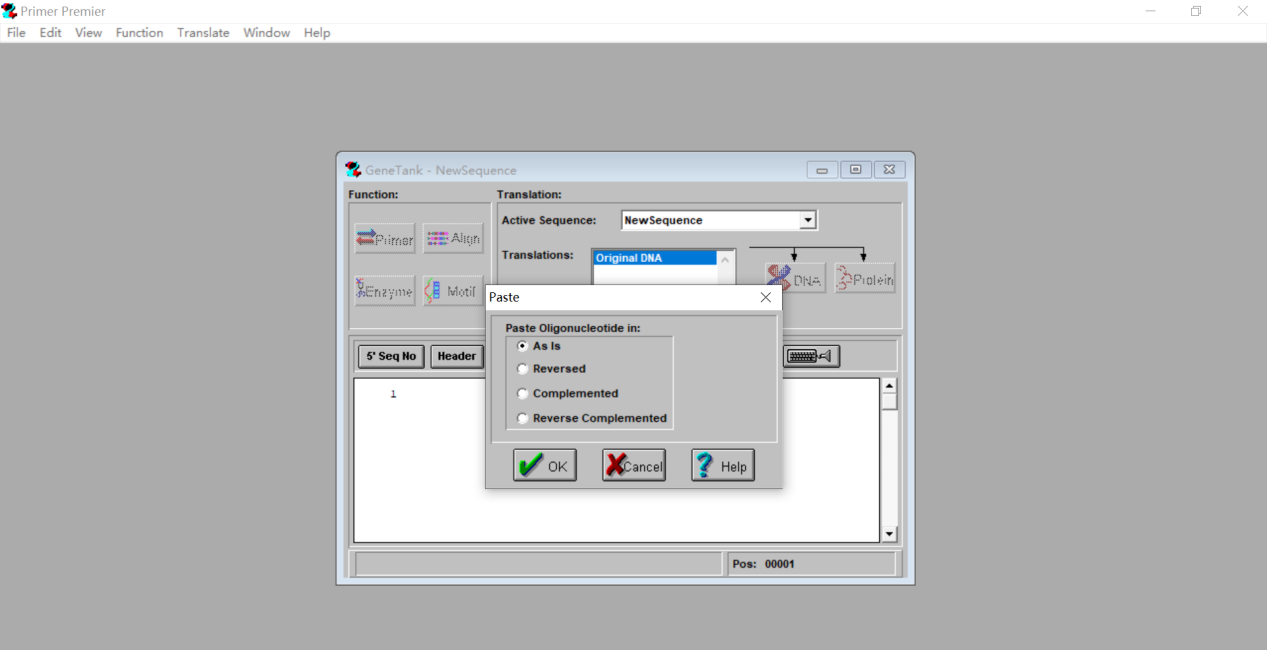
3.5 点击空白处粘贴我们之前找好的目的基因的CDS序列,选择As is。

3.6 点击左上角的Primer,S为正向引物,A为反向引物
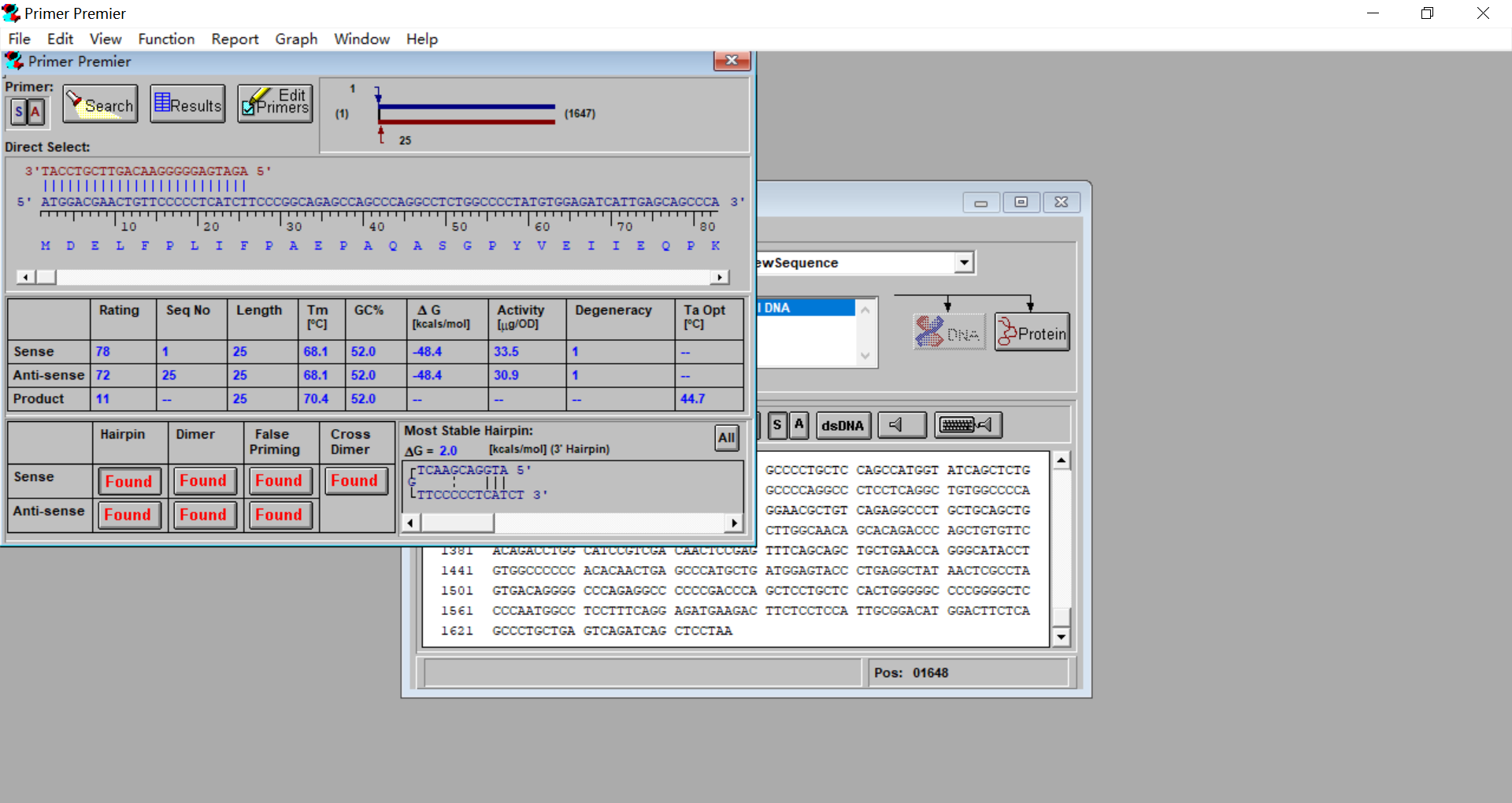

3.7 点击Edit Primers编辑序列,可调整引物长短,添加酶切位点序列以及保护碱基。如果显示Hairpin,Dimer,False Priming,Cross Dimer或者Tm值太高或太低,则需要调整。

如果检测引物特异性
3.8 NCBI主页最下端找到Primer-BLAST,打开;

3.9 输入上下游引物序列,点击最下方 ;
;

3.10 如果只能检测到你的目的蛋白基因,则表明特异性较好。
拿到合成的引物之后,我们就可以进行目的基因的扩增了。合成目的基因通常是以目标物种的基因组为模板。
由于真核生物的基因组中存在内含子序列,在mRNA的剪接加工过程中被切除,因此,在构建真核基因克隆时基因组DNA不能直接用作PCR扩增的模板,只能从细胞或者组织中提取总RNA或者mRNA,反转录cDNA作为模板通过PCR扩增目的基因。
4、从细胞中提取总RNA
4.1 从细胞培养箱中取出已经长满细胞的细胞培养皿,小心吸出培养基,加入4ml预冷PBS,倾斜细胞培养皿3次以充分洗涤细胞,然后小心吸尽PBS溶液。
4.2 培养皿中加入1ml Trizol试剂裂解细胞,冰上静置5min,枪头吹打,室温静置5min;
4.3 将细胞裂解液吸到1.5ml EP管中,加入氯仿0.2 ml,震荡仪震荡15s。
4.4 室温静置2-3min,12000xg,4℃,15min离心。
4.5 离心后液体分为三层(上层无色为RNA,中层为DNA,底层为蛋白质),小心吸取上层无色液体至新的EP管中。
4.6 加入等体积异丙醇,混匀,冰上静置10min,12000xg,4℃,10min离心。
4.7 此时在管底可见RNA白色沉淀,弃去上清。
4.8 加入75%乙醇1ml,震荡仪震荡30s,7500xg,4℃,5min离心。
4.9 小心去上清,管内沉淀在超净台中鼓风静置干燥3-5min。
4.10 加入20ul DEPC水溶解。水浴或者加热器55-60℃,10-15min。
4.11 测定纯度和浓度。吸光度A260/280比值接近2表明纯度较高。
部分相关产品
| 货号 | 名称 | 规格 | 用途 |
| abs9331 | RNA提取试剂盒 | 100ml | RNA提取试剂盒 |
| 25050070 | Amersham RNAspin Mini Kit | 20T | RNA提取试剂盒 |
5、反转录获得cDNA
反转录通常使用试剂盒和PCR仪器进行操作,这里参照了爱必信的RT-PCR Kit产品说明书进行介绍。
产品组分:

实验方法:
5.1 将 RNA 模板、引物、2×One-Step RT-PCR SuperMix,RT Enzyme Mix 和RNase Free Water 溶解并置于冰上备用。
5.2 配置以下反应体系:

5.3 轻轻混匀后短暂离心,使管壁上的溶液收集到管底。
5.4 常用反应程序:
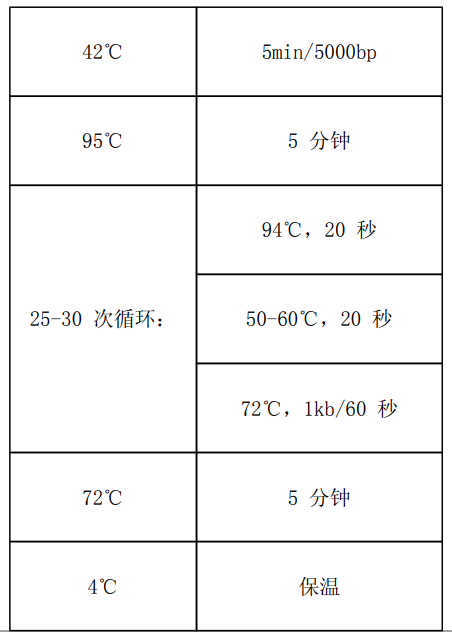
5.5 反应完成后,测定纯度和浓度。
部分相关产品
| 货号 | 名称 | 规格 | 用途 |
| abs60076 | 反转录试剂盒 | 50T | 反转录试剂盒 |
| 27925901 | Amersham Ready-To-Go RT-PCR Beads | 0.2ml | 反转录试剂盒 |
6、目的基因的扩增
目的基因的扩增通常也是使用试剂盒和PCR仪器完成。这里参照了爱必信的2xPfu Master Mix产品说明书进行介绍。
注:所有组分应仔细混匀并离心后开启,所有 PCR操作过程应在冰上进行。
操作示例:以50 μl PCR反应体系为例 。
6.1 按照下表配制 PCR反应体系

*模板量:1-2 μl RT-PCR反应后的 cDNA。
6.2 PCR反应循环的设置

6.3 结果检测:取 2-5 μl反应液电泳观察结果,测定纯度和浓度。
目的基因扩增之后,就要将目的基因和表达载体连接在一起。通常使用酶切连接的方法,将目的基因和表达载体分别用同样的两个限制性内切酶进行双酶切,得到互补的酶切位点,然后用T4连接酶将这两部分连接在一起。目的基因扩增之后,目的基因存在的缓冲体系可能会影响到酶切效果,所以通常先进行目的基因的纯化,也叫作切胶回收。
部分相关产品
| 货号 | 名称 | 规格 | 用途 |
| abs60057 | 2×HotStart Taq PCR Mix | 5×1ml | 目的片段DNA扩增 |
| abs60055 | 2×Pfu PCR MasterMix | 5×1ml | 目的片段DNA扩增 |
7、目的基因的纯化
先进行跑胶,然后进行切胶回收。切胶回收通常也使用试剂盒进行操作,这里参照爱必信的DNA Gel/PCR Purification Kit产品说明书进行介绍。
7.1 取50xTAE缓冲液20ml加水至1000ml,配制成1xTAE电泳缓冲液,备用。
7.2 称取0.5g琼脂糖,置于200ml锥形瓶中,加入50ml 1xTAE电泳缓冲液,在微波炉里加热至琼脂糖全部融化,取出摇匀。加热过程中要不时摇动,使琼脂糖在溶液中分散均匀。
7.3 在制胶槽中插好梳子,向冷却的琼脂糖中加入GelRED染色剂,缓慢倒入制胶槽中。把制胶槽放入电泳仪中,在电泳仪中倒入TAE电泳缓冲液。
7.4 将50ul PCR产物中加入10ul 6x loading buffer并混匀,用移液器小心加入到加样孔中。在旁边的孔中加入核酸Marker。
7.5 加样完成后,盖上电泳仪盖子,启动电源。电压通常设置为100V,当溴酚蓝指示跑到适合位置时,关闭电源停止电泳。
7.6 将凝胶转移至核酸检测仪中,在紫外灯下快速准确的切下含有目的DNA片段的琼脂糖凝胶块,放进1.5ml离心管中。
以下为爱必信DNA Gel/PCR Purification Kit产品说明书进行胶回收介绍。
产品组分:

7.7 DNA吸附柱平衡处理:向Gel Recovery Column中加入200μl buffer CBS,12000rpm离心1min,倒掉收集管中的废液,将Gel Recovery Column重新放回到收集管中。再向Gel Recovery Column中加入200μl的ddH2O,12000rpm离心1min,倒掉收集管中的废液。将Gel Recovery Column重新放回到收集管中。
7.8 从琼脂糖凝胶中切下含有目的片段的凝胶,估计重量或精确称量重量。每100mg 1%琼脂糖凝胶加入100μl Binding Solution。
7.9 于50-60℃水浴5-10min,期间每2-3min间断轻微颠倒混匀,直至胶块完全融化。
7.10 于50-60℃水浴5-10min,期间每2-3min间断轻微颠倒混匀,直至胶块完全融化。
7.11 将吸附柱重新放回收集管中,加入500μl WA Solution,于12000rpm离心1min,倒掉收集管中废液。
7.12 将吸附柱重新放回收集管中,加入500μl Wash Solution,于12000rpm离心1min,倒掉收集管中废液。
7.13 重复上个步骤一次。
7.14 将吸附柱重新放回收集管中,12000rpm离心1min,打开吸附柱盖子,室温放置5-10min或50℃放置3-5min,以彻底去除Wash Solution。
7.15 将吸附柱放入干净的1.5ml收集管中(试剂盒自带),对于膜中央悬空加入30-50μl Elution Buffer,盖好盖子,37℃放置2min,12000rpm离心1min,离心管中的液体即为包含目的基因的溶液。
7.16 检测纯度及浓度。
部分相关产品
| 货号 | 名称 | 规格 | 用途 |
| abs60098 | PCR & DNA Fragment Purification Kit | 50T | DNA片段胶回收试剂盒 |
| 28104 | QIAquick PCR Purification Kit | 50T | DNA片段胶回收试剂盒 |
8、目的基因和表达载体的双酶切
酶切操作建议参考购买的限制性内切酶的使用说明书,这里介绍常规方法。
8.1 在两个PCR管中分别混合以下成分:
表达载体酶切:
| 表达载体 | 2ug |
| 10x缓冲液 | 5ul |
| 酶1 | 1ul |
| 酶2 | 1ul |
| ddH2O | 补齐至30ul |
PCR产物酶切:
| 目的片段 | 43ul |
| 10x缓冲液 | 5ul |
| 酶1 | 1ul |
| 酶2 | 1ul |
8.2 轻弹管壁混匀,短暂离心。
8.3 放入PCR仪中,37℃,3h。
8.4 跑电泳进行胶回收。
9、目的片段和表达载体的连接
9.1 取PCR管,加入酶切后的表达载体片段2ul,酶切后的目的基因片段6ul,T4 DNA连接酶1ul,10* T4 buffer 1ul,轻弹管壁混匀,短暂离心。
9.2 放入PCR仪中,16℃过夜。
部分相关产品
| 货号 | 名称 | 规格 | 用途 |
| 28954549 | PGEX-4T-1 | 25ug | 表达载体 |
| 28954648 | PGEX-6P-1 VECTOR | 25ug | 表达载体 |
| abs60084 | T4 DNA Ligase | 500U | 连接酶 |
| abs60085 | T-Vector快速克隆试剂盒 | 20T | 常规克隆 |
| abs60089 | pBM21快速克隆试剂盒 | 20T/3×20T | 常规克隆 |
| abs60091 | T-Vector pTOPO快速克隆试剂盒 | 20T/100T | TOPO克隆 |
| abs60095 | pBM16A Toposmart快速克隆试剂盒 | 20T/3×20T | TOPO克隆 |
| abs9314 | 核酸预制胶 | 10片/盒 | 核酸预制胶 |
| abs60002 | DL 2000 DNA Marker | 100T(250ul×2支) | DNA Marker |
10、转化
目的基因与载体的连接反应中,存在多种可能的连接方式,除了正确连接外,也可能出现载体和载体连接,目的基因和目的基因连接,或者部分连接的情况,因此需要把正确连接的重组质粒筛选出来。通常将连接产物转化到大肠杆菌中,通过菌落PCR或者酶切鉴定的方法筛选出阳性克隆株,然后送到测序公司进行测序。测序准确的阳性克隆株就可以进行大量扩增和保存。
常用的大肠杆菌感受态细胞为DH5α或者TOP10。
10.1 取出感受态大肠杆菌,放于冰上使其慢慢融化。
10.2 将连接产物加入100ul感受态大肠杆菌中,轻轻混匀,冰上孵育30分钟。
10.3 将EP管放入42℃水浴箱中热激90秒,再迅速放于冰上2分钟。
10.4 EP管中加入400u LB培养基,置于摇床上,37℃,500rpm培养45分钟。
部分相关产品
| 货号 | 名称 | 规格 | 用途 |
| abs60101 | DH5α 感受态细胞 | 10×100ul/20×100ul | 感受态细胞 |
11、涂板培养
500rpm离心1-2分钟,留100ul培养基上清,将转化后的大肠杆菌轻轻混匀,涂于含抗生素的LB固体培养基上,37℃恒温培养箱中倒置培养过夜。
12、挑克隆及阳性克隆株鉴定
挑取3-5个单克隆分别放入不同的离心管中(如果菌落不多可以全部挑取),此时可以进行菌落PCR和酶切鉴定。
1)菌落PCR
同质粒构建时的PCR扩增,使用同样的引物,模板换成挑出的菌落;跑胶如果有扩增的条带则为阳性克隆株;
2)酶切鉴定
菌落提质粒,使用质粒构建时的酶进行酶切;跑胶如果看到目的片段则为阳性克隆株;
部分相关产品
| 货号 | 名称 | 规格 | 用途 |
| abs9224 | Ampicillin Sodium | 5g/25g/100g | 氨苄抗生素 |
| abs60057 | 2×HotStart Taq PCR Mix | 5×1ml | 目的片段DNA扩增 |
| abs60055 | 2×Pfu PCR MasterMix | 5×1ml | 目的片段DNA扩增 |
| abs9314 | 核酸预制胶 | 10片/盒 | 核酸预制胶 |
| abs60002 | DL 2000 DNA Marker | 100T(250ul×2支) | DNA Marker |
13、质粒提取
酶切鉴定需要提质粒,如果直接送公司测序,此步也可省略。提取质粒一般也是使用试剂盒进行操作,这里参照爱必信的质粒小量快速提取试剂盒产品说明书进行介绍。
产品组份:

实验方法:
13.1 向吸附柱AC 中(吸附柱放入收集管中)加 500μl 的平衡液 BL,12,000rpm 离心1min, 倒掉收集管中的废液,将吸附柱重新放回收集管中。
13.2 取 1.5-4.5 毫升过夜培养的菌液 12,000rpm 离心 30sec,尽可能的倒干上清,收集菌体。
13.3 用 250μl 溶液 P1 重悬菌体沉淀,涡旋振荡至彻底悬浮。
13.4 加 250μl 溶液 P2,温和地上下翻转 4 -7 次(裂解时间不超过 5min)使菌体充分裂解。
13.5 加 350μl 溶液 P3,立即温和地上下翻转 4 -7 次,充分混匀,此时会出现白色絮状沉淀,12,000rpm 离心 5 min。
13.6 将上清加入到过滤柱E中(过滤柱放入 1.5ml或2ml离心管中),12,000rpm离心2 min,收集液体。如上清量较大,请分两次离心。
13.7 将上述液体转入吸附柱 AC 中(吸附柱放入收集管中),12,000rpm 离心 30-60 sec,倒掉管中的废液。 可选步骤: 所用菌株为 JM 系列、HB101 等 endA 菌株或野生型菌株时,由于核酸酶含量丰富, 应加入 500μl 去蛋白液 PD,12,000rpm 离心 30-60 sec,弃废液。
13.8 加入 500μl 漂洗液 WB(请先检查是否已加入无水乙醇!),12,000rpm 离心 30-60sec 弃掉废液。
13.9 重复上个步骤。
13.10 将吸附柱 AC 放回空收集管中,12,000rpm 离心 2 min,将吸附柱置于室温放置数min,以彻底晾干吸附材料中残余的漂洗液, 以免漂洗液中残留乙醇抑制下游反应。
13.11 取出吸附柱 AC,放入一个干净的离心管中,室温放几分钟。
13.12 在吸附膜的中间部位加 50μl-100μl 洗脱缓冲液 EB(洗脱缓冲液事先在 65-70℃水浴中加热效果更好),室温放置 2 min,12,000rpm 离心 1 min。如果需要较多量质粒,可将得到的溶液重新加入离心吸附柱中,离心 1 min。(注意:若用 ddH2O 做洗脱液,应保证其 pH 值在 7.0-8.5 范围内,pH 值低于 7.0 会降低洗脱效率。洗脱缓冲液体积不应少于 50 μl,体积过小影响回收效率。且 DNA 产物应保存在-20℃,以防 DNA 降解。)
部分相关产品
| 货号 | 名称 | 规格 | 用途 |
| abs60023 | 质粒小量快速提取试剂盒 | 50T | 质粒提取试剂盒 |
| 12943 | Plasmid Plus Midi Kit | 25T | 质粒提取试剂盒 |
| 12243 | QIAfilter Plasmid Midi Kit | 25T | 质粒提取试剂盒 |
14、测序
鉴定后的阳性克隆株进行培养,送公司测序。测序成功的菌株进行培养和保存。
15、目的蛋白的表达
目的蛋白表达通常使用293细胞或者CHO细胞。将鉴定成功后的克隆株培养之后,提取重组质粒。然后将重组质粒转染到细胞中。提质粒可参考前文步骤。
15.1 细胞复苏
在无菌超净台中预备培养瓶,加5ml预热培养基,盖好瓶盖待用。将细胞从液氮中取出,置于37℃水浴中迅速解冻,用酒精棉球擦拭后移至超净工作台,(如果需要去除冷冻保护剂,例如DMSO或Glycerol,则先800rpm离心3min,吸出冷冻保护剂),加入少量培养基吹打并混匀细胞,吸入至培养瓶中。
15.2 细胞传代
1、取出细胞培养瓶,吸出培养基,加1ml PBS润洗细胞,吸出PBS丢弃;
2、加入1ml胰酶,轻轻晃动培养瓶使浸润所有细胞,放入37℃培养箱中;
3、加入2ml含血清的培养基终止消化,吹打细胞使细胞脱落;
4、收集细胞悬液至离心管中,1200rpm离心3min,吸出上清;
5、加入新鲜培养基,吹打混匀,分别加入到两个培养瓶中。
注:如果是悬浮细胞,则无需消化步骤。
15.3 细胞计数
1、使用胰酶消化细胞,加入含血清的培养基终止消化。
2、如果需要查看细胞活性,可使用台盼蓝染色。取180ul的细胞液至EP管中,加入20ul台盼蓝母液(4%台盼蓝),用移液器吹打混匀;
3、取10ul含有台盼蓝的细胞液加入细胞计数器中,使用显微镜进行细胞计数。
15.4 细胞转染
细胞转染常用的方法有脂质体转染法,电穿孔转染法,病毒转染法等等,这里以Polyplus的JetPRIME转染试剂为例进行介绍。
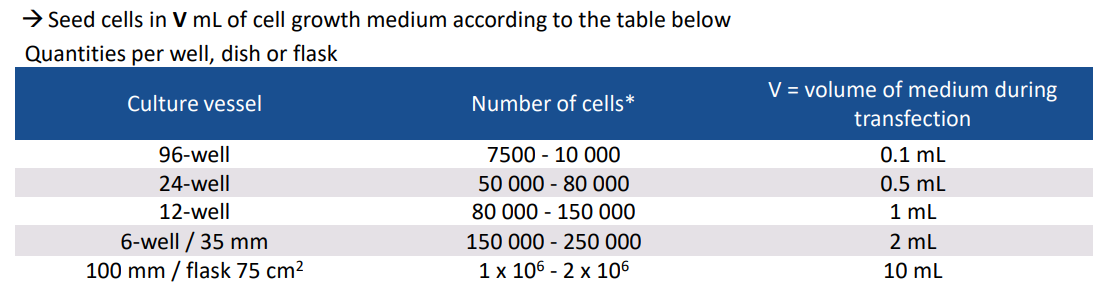
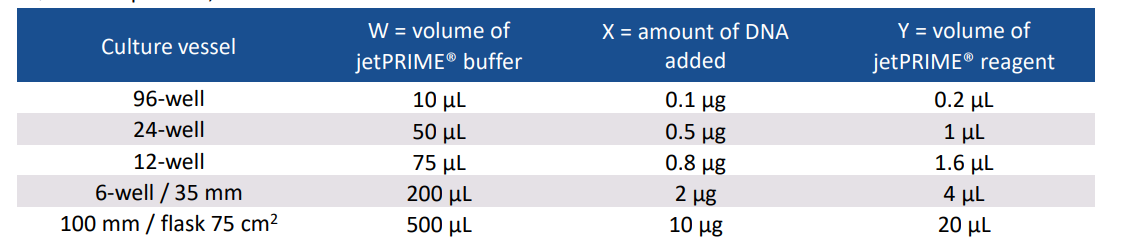
1、为了达到最好的转染状态,一般选择细胞融合度为60%-80%时进行转染。推荐的细胞数可以参考以下表格:
2、稀释X ug的质粒至W ul的JetPRIME缓冲液中,震荡10s,瞬离;
3、加入Y ul JetPRIME试剂至上一步的混合物中;
4、震荡1s,瞬离,室温孵育10min;
5、把转染混合物加至细胞培养瓶中。
具体体积参考下表:
15.5 蛋白检测
取细胞加裂解液进行细胞裂解,取部分上清和6xloading buffer混合,在95℃加热5min。使用SDS-PAGE进行跑胶并用考马斯亮蓝 R250进行染色,检测蛋白表达。
部分相关产品
| 货号 | 名称 | 规格 | 用途 | |
| 培养基 | 12-770Q | PowerCHO-1 serum-free medium 1L | 1L | 无血清和非动物来源培养基 |
| BEBP12-932Q | eCHO Feed Medium - 1L bottle | 1L | CHO细胞培养基 | |
| BEBP12-764Q | Pro293 a Serum-free Medium – Chemically Defined for 293 Adherent Cells | 1L | 用于293贴壁细胞培养,重组蛋白生产 | |
| BEBP02-030Q | ProVero 1 Serum-free Medium | 1L | 培养基,用于MDCK和vero细胞的生长 | |
| 转染试剂 | 301105 | PolyFect Transfection Reagent | 1ml | 细胞转染试剂 |
| 116-001 | FectoPRO® | 1ml | 蛋白和抗体生产专用,悬浮的293、CHO细胞转染 | |
| 114-01 | jetPRIME® | 1ml | 通用型转染试剂,适用于贴壁细胞 | |
| AAB-1001 | Nucleofector™ 2b Device | / | 电转仪 | |
| V4XP-3024 | P3 Primary Cell 4D X Kit L | 100ul | 电转试剂盒 | |
| 336921 | SureENTRY Transduction Regent | 1EA | 慢病毒和逆转录病毒转染试剂 | |
| 裂解液 | abs9229 | RIPA裂解液 | 100ml | 细胞裂解液 |
| 28941279 | Mammalian Cell Lysis Buffer | 500ml | 哺乳动物细胞裂解液 | |
| 染色液 | abs964 | 考马斯亮蓝染色试剂盒(常规型)R250 | 100ml | SDS-PAGE蛋白电泳凝胶染色 |
16、蛋白纯化
成功表达目标蛋白的细胞经过裂解,超声和过滤后可开始进行蛋白纯化。标签蛋白纯化通常使用2步或者3步纯化方式。
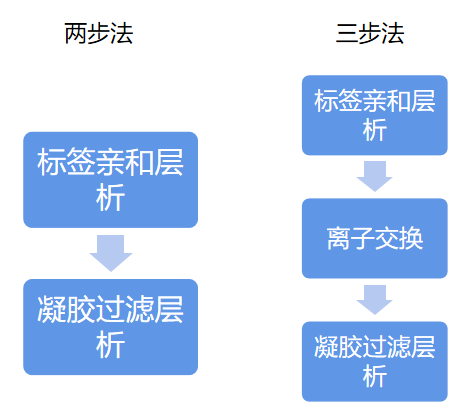
16.1 标签亲和层析
根据您构建质粒时选择的标签选择对应的标签亲和层析产品。常用的标签有HIS,GST,MBP,Strep,FLAG等等,这里以His标签预装柱和重力柱为例进行介绍。
(一) HisTrap HP 预装柱使用说明
结合缓冲液: 20mM 磷酸钠缓冲液, 0.5M 氯化钠, 20-40mM 咪唑, PH7.4
洗脱缓冲液: 20mM 磷酸钠缓冲液, 0.5M 氯化钠, 500mM 咪唑, PH7.4
样品上样前需要过滤,缓冲液需要预先过滤和超声去除气泡。
1、 系统设置一个低流速, 移除预装柱顶部的堵头,把柱子连接到 ӒKTA 纯化仪的接头上,注意要液滴对液滴进行连接,防止引入气泡。
2、 去除柱子底部的堵头,连接到纯化仪上。
3、 用 3-5 个柱体积的蒸馏水洗去乙醇。
4、 用 5 个柱体积的结合缓冲液平衡柱子,建议流速是 1ml/min(1ml 体积柱子)和 5ml/min(5ml 体积柱子)。
5、 用上样环或者 superloop 加入预处理的样品(样品在上柱前一定要进行离心和过滤),在上样时建议流速为 0.2-1ml/min(1ml 体积柱子)和 0.5-5ml/min(5ml 体积柱子)。
6、 用结合缓冲液洗涤至少 10 到 15 个柱体积,直到吸收峰达到稳定基线或在流出物中没有物质流出。洗涤过程中建议保持流速为 1-2ml/min(1ml 体积柱子)和 5-10ml/min(5ml体积柱子)。
7、 用洗脱缓冲液采用一步洗脱或者线性梯度洗脱(拉咪唑浓度梯度)。一步洗脱通常 5 个柱体积,线性洗脱通常 10-20 个柱体积。在洗脱过程中保持流速为 1-2ml/min(1ml 体积柱子)和 5-10ml/min(5ml 体积柱子)。
8、 洗脱后,用 3-5 个柱体积的结合缓冲液洗涤柱子,然后加入 20%乙醇。拧上柱子上下的堵头,防止柱子变干。
Note:如果需要去除纯化后样品中的咪唑,建议使用 HiTrap Desalting 脱盐柱或者 PD-10 Dealting 脱盐柱。
(二) Ni Sepharose 6 FF 装重力柱使用说明
结合缓冲液: 20mM 磷酸钠缓冲液, 0.5M 氯化钠, 20-40mM 咪唑, PH7.4
洗脱缓冲液: 20mM 磷酸钠缓冲液, 0.5M 氯化钠, 500mM 咪唑, PH7.4
样品上样前需要过滤,缓冲液需要预先过滤和超声去除气泡。
一、 准备 PD-10 空柱
1、 用 20%的乙醇洗涤滤膜。
2、 用蒸馏水润洗滤膜。
3、 将滤膜放入 PD-10 空柱。
二、 填料准备
1、 轻柔震荡瓶子,直到填料悬浊液均一。
2、 将需要量的填料悬浊液从瓶中转移到离心管中。
3、 用 500xg 离心 5min 以沉淀填料。
4、 除去上清,加入适量蒸馏水。
5、 轻柔的振荡填料悬浊液 3min, 用 500xg 离心 5min。
6、 除去上清,加入适量结合缓冲液,重复步骤 5.
7、 把填料悬浊液转移到量筒中。
8、 加入适量体积的结合缓冲液,使悬浊液中的填料浓度达到 50%。
三、 重力柱纯化
1、 将样品加入含有 50%填料的悬浊液中(样品在上柱前要进行离心和过滤)。 Ni Sepharose 6FF 的平均载量是 40mg/ml。则 1ml 的 50%悬浊液的载量为大约 20mg 蛋白。
2、 将样品和填料混合物在摇床上低速混合 1h。
3、 将样品和填料混合物加入到 PD-10 的空柱中,收集流出物。
4、 用结合缓冲液洗涤 2-5 个柱体积,收集流出物。
5、 用 4 个柱体积的洗脱缓冲液洗脱,收集流出物。
| 货号 | 品名 | 规格 | 用途 | |
| 预装柱 | 29051021 | HisTrap HP | 1 × 1 ml | His标签蛋白纯化 |
| 17524701 | HisTrap HP | 5 × 1 ml | His标签蛋白纯化 | |
| 17524801 | HisTrap HP | 1 × 5 ml | His标签蛋白纯化 | |
| 17524802 | HisTrap HP | 5 × 5 ml | His标签蛋白纯化 | |
| 17531901 | HisTrap FF | 5 × 1 ml | His标签蛋白纯化 | |
| 17525501 | HisTrap FF | 5 × 5 ml | His标签蛋白纯化 | |
| 29048586 | HisTrap excel | 1 x 1 ml | His标签蛋白纯化 | |
| 17371205 | HisTrap excel | 5 x 1 ml | His标签蛋白纯化 | |
| 17371206 | HisTrap excel | 5 x 5 ml | His标签蛋白纯化 | |
| 29048631 | HisTrap FF Crude | 1 × 1 ml | His标签蛋白纯化 | |
| 11000458 | HisTrap FF Crude | 5 × 1 ml | His标签蛋白纯化 | |
| 17528601 | HisTrap FF Crude | 5 × 5 ml | His标签蛋白纯化 | |
| 填料 | 17526801 | Ni Sepharose HP | 25 ml | His标签蛋白纯化 |
| 17526801 | Ni Sepharose HP | 25 ml | His标签蛋白纯化 | |
| 17526802 | Ni Sepharose HP | 100 ml | His标签蛋白纯化 | |
| 17531806 | Ni Sepharose 6 FF | 5 ml | His标签蛋白纯化 | |
| 17531801 | Ni Sepharose 6 FF | 25 ml | His标签蛋白纯化 | |
| 17531802 | Ni Sepharose 6 FF | 100 ml | His标签蛋白纯化 | |
| 17531803 | Ni Sepharose 6 FF | 500 ml | His标签蛋白纯化 | |
| 17371201 | Ni Sepharose excel | 25ml | His标签蛋白纯化 | |
| 28967390 | His Mag Sepharose Ni | 5x1ml | His标签磁珠 | |
| 28967388 | His Mag Sepharose Ni | 2x1 ml | His标签磁珠 |
GST标签蛋白纯化相关产品
| 货号 | 品名 | 规格 | 用途 | |
| 预装柱 | 17528101 | GSTrap HP | 5 × 1 ml | GST标签蛋白纯化 |
| 17528201 | GSTrap HP | 1 × 5 ml | GST标签蛋白纯化 | |
| 17528202 | GSTrap HP | 5 × 5 ml | GST标签蛋白纯化 | |
| 17513001 | GSTrap FF | 5 × 1 ml | GST标签蛋白纯化 | |
| 17513101 | GSTrap FF | 1 × 5ml | GST标签蛋白纯化 | |
| 17513102 | GSTrap FF | 5 × 5ml | GST标签蛋白纯化 | |
| 29048609 | GSTrap 4B | 1 × 1 ml | GST标签蛋白纯化 | |
| 28401747 | GSTrap 4B | 1 × 5 ml | GST标签蛋白纯化 | |
| 填料 | 17527901 | Glutathione Sepharose HP | 25 ml | GST标签蛋白纯化 |
| 17527902 | Glutathione Sepharose HP | 100 ml | GST标签蛋白纯化 | |
| 17513201 | Glutathione Sepharose 4 FF | 25 ml | GST标签蛋白纯化 | |
| 17513202 | Glutathione Sepharose 4 FF | 100 ml | GST标签蛋白纯化 | |
| 17075601 | Glutathione Sepharose 4B | 10 ml | GST标签蛋白纯化 | |
| 17075605 | Glutathione Sepharose 4B | 100 ml | GST标签蛋白纯化 | |
| 切除酶 | 27084301 | PreScission Protease | 500 units | GST标签切除酶 |
| 27084601 | Thrombin Protease | 500 units | GST标签切除酶 |
Strep标签蛋白纯化相关产品
| 货号 | 品名 | 规格 | 用途 | |
| 预装柱 | 29401317 | StrepTrap XT | 1 × 1 ml | Strep标签蛋白纯化 |
| 29401320 | StrepTrap XT | 5 × 1 ml | Strep标签蛋白纯化 | |
| 29401322 | StrepTrap XT | 1 × 5 ml | Strep标签蛋白纯化 | |
| 29401323 | StrepTrap XT | 5 × 5 ml | Strep标签蛋白纯化 | |
| 填料 | 29401324 | Strep-Tactin XT Sepharose | 10ml | Strep标签蛋白纯化 |
| 29401326 | Strep-Tactin XT Sepharose | 50ml | Strep标签蛋白纯化 |
MBP标签蛋白纯化相关产品
| 货号 | 品名 | 规格 | 用途 | |
| 预装柱 | 28918779 | MBPTrap HP | 1 × 5 ml | MBP标签蛋白纯化 |
| 29048641 | MBPTrap HP | 1 × 1 ml | MBP标签蛋白纯化 | |
| 28918778 | MBPTrap HP | 5 × 1 ml | MBP标签蛋白纯化 | |
| 28918780 | MBPTrap HP | 5 × 5 ml | MBP标签蛋白纯化 | |
| 填料 | 28935597 | Dextrin Sepharose HP | 25 mL | MBP标签蛋白纯化 |
| 28935598 | Dextrin Sepharose HP | 100 mL | MBP标签蛋白纯化 |
16.2 离子交换
离子交换需要根据您蛋白的等电点选择阴离子交换柱或者阳离子交换柱。
1、如果您的蛋白的等电点小于缓冲液的PH,则选择阴离子交换柱(Q,DEAE);
2、如果您的蛋白的等电点大于缓冲液的PH,则选择阳离子交换柱(S,SP,CM);
这里以Q柱为例进行介绍。
结合缓冲液:缓冲液的PH比目标蛋白的等电点高一个PH。
推荐缓冲液:

洗脱缓冲液:结合缓冲液+1M NaCl
样品上样前需要过滤,置换到结合缓冲液中。缓冲液需要预先过滤和超声去除气泡。
1、 系统设置一个低流速, 移除预装柱顶部的堵头,把柱子连接到 ӒKTA 纯化仪的接头上,注意要液滴对液滴进行连接,防止引入气泡。
2、 去除柱子底部的堵头,连接到纯化仪上。
3、 用 3-5 个柱体积的蒸馏水洗去乙醇。
4、 用 5 个柱体积的结合缓冲液平衡柱子,建议流速是 1ml/min(1ml 体积柱子)和 5ml/min(5ml 体积柱子)。
5、 用上样环或者 superloop 加入预处理的样品(样品在上柱前一定要进行离心和过滤),在上样时建议流速为 1ml/min(1ml 体积柱子)和 5ml/min(5ml 体积柱子)。
6、 用结合缓冲液洗涤至少 5个柱体积,直到吸收峰达到稳定基线或在流出物中没有物质流出。洗涤过程中建议保持流速为 1ml/min(1ml 体积柱子)和 5ml/min(5ml体积柱子)。
7、 用洗脱缓冲液采用一步洗脱或者线性梯度洗脱(拉NaCl梯度)。一步洗脱通常 5-10 个柱体积,线性洗脱通常 10-20 个柱体积。在洗脱过程中保持流速为 1ml/min(1ml 体积柱子)和 5ml/min(5ml 体积柱子)。
8、 洗脱后,使用5个柱体积结合缓冲液+1M NaCl进行再生,然后用 5-10 个柱体积的结合缓冲液洗涤柱子,然后加入 20%乙醇。拧上柱子上下的堵头,防止柱子变干。
| 货号 | 品名 | 规格 | 用途 | |
| 预装柱 | 28934388 | HiTrap Capto IEX Selection Kit | 5根不同柱子 | 阴、阳离子不同配基预装柱 |
| 29275878 | Capto HiRes Q 5/50 | 5 x 50mm | 高分辨率阴离子交换柱 | |
| 29275881 | Capto HiRes Q 10/100 | 10 x 100mm | 高分辨率阴离子交换柱 | |
| 29275877 | Capto HiRes S 5/50 | 5 x 50mm | 高分辨率阳离子交换柱 | |
| 29275879 | Capto HiRes s 10/100 | 10 x 100mm | 高分辨率阳离子交换柱 | |
| 29051325 | HiTrap Q HP | 1 × 1 ml | 阴离子交换柱 | |
| 17115301 | HiTrap Q HP | 5 × 1 ml | 阴离子交换柱 | |
| 17515601 | HiTrap Q FF | 5 × 5 ml | 阴离子交换柱 | |
| 11001302 | HITRAP CAPTO Q, 5X1ML | 5 × 1 ml | 阴离子交换柱 | |
| 11001303 | HiTrap Capto Q | 5 × 5 ml | 阴离子交换柱 | |
| 29051324 | HiTrap SP HP | 1 × 1 ml | 阳离子交换柱 | |
| 17115101 | HiTrap SP HP | 5 × 1 ml | 阳离子交换柱 | |
| 17115201 | HiTrap SP HP | 5 × 5 ml | 阳离子交换柱 | |
| 17505401 | HiTrap SP FF | 5 × 1 ml | 阳离子交换柱 | |
| 17515701 | HiTrap SP FF | 5 × 5 ml | 阳离子交换柱 | |
| 17544122 | HITRAP CAPTO S, 5X1 ML | 5 × 1 ml | 阳离子交换柱 | |
| 17544123 | HiTrap Capto S | 5 × 5 ml | 阳离子交换柱 | |
| 17505501 | HiTrap DEAE FF | 5 × 1 ml | 弱阴离子交换柱 | |
| 17515401 | HiTrap DEAE FF | 5 × 5 ml | 弱阴离子交换柱 | |
| 28916537 | HiTrap Capto DEAE, 5X1ML | 5 × 1 ml | 弱阴离子交换柱 | |
| 28916540 | HiTrap Capto DEAE | 5 × 5 ml | 弱阴离子交换柱 | |
| 17515501 | HiTrap CM FF | 5 × 5 ml | 弱阳离子交换柱 | |
| 28405846 | HiTrap Capto adhere | 5 × 5 ml | 阴离子交换柱 | |
| 填料 | 17531610 | CAPTO Q, 25 ML | 25 ml | 阴离子交换填料 |
| 17531602 | Capto Q 100 ml | 100 ml | 阴离子交换填料 | |
| 17051010 | Q Sepharose FF | 25 ml | 阴离子交换填料 | |
| 17051001 | Q Sepharose FF | 300 ml | 阴离子交换填料 | |
| 17544110 | CAPTO S, 25 ML | 25 ml | 阳离子交换填料 | |
| 17544101 | CAPTO S, 100 ML | 100 ml | 阳离子交换填料 | |
| 17072910 | SP Sepharose FF | 25 ml | 阳离子交换填料 | |
| 17072901 | SP Sepharose FF | 300 ml | 阳离子交换填料 | |
| 17544301 | CAPTO DEAE, 100 ML | 100 ml | 阴离子交换填料 | |
| 17070910 | DEAE Sepharose FF | 25 ml | 阴离子交换填料 | |
| 17070901 | DEAE Sepharose FF | 500 ml | 阴离子交换填料 | |
| 17071910 | CM Sepharose FF | 25 ml | 阳离子交换填料 | |
| 17071901 | CM Sepharose FF | 500 ml | 阳离子交换填料 | |
| 17544410 | Capto adhere | 25 ml | 多模式填料 |
16.3 凝胶过滤层析
凝胶过滤层析主要用于精细纯化或者去除多聚体。凝胶过滤层析主要根据上样量和蛋白分子量进行选择。
这里以Superdex 200 Increase 10/300为例进行介绍。
缓冲液的选择:
缓冲液可以根据您的样品或者下游应用进行选择,由于在极低盐浓度的情况下,酸性和碱性蛋白质都可能发生离子相互作用,因此推荐的缓冲液是 0.01 至 0.05 M 磷酸钠再加上0.15 M NaCl,pH 7.4。
样品处理:
分子量:10KD-600KD
上样体积:25-500ul
蛋白浓度:样品中最多50 mg/mL,在低于10 mg/mL时可获得更高的分辨率。
准备:将样品溶解在缓冲液中,10 000 g 离心10 分钟,用0.22 μm 过滤器过滤。
1、根据层析柱的耐压在系统中设置柱前压和delta压
2、系统设置一个低流速, 移除预装柱顶部的堵头,把柱子连接到 ӒKTA 纯化仪的接头上,
注意要液滴对液滴进行连接,防止引入气泡。
3、去除柱子底部的堵头,连接到纯化仪上。
4、以0.75 mL/min 的流速使用至少 2 个柱体积 (CV) 的室温去离子水进行平衡。
5、以0.75 mL/min 的流速用至少 2 个柱体积 (CV) 缓冲液平衡层析柱。
6、以0.75 mL/min 的流速进行上样和缓冲液洗脱。
7、纯化结束后,用2CV的去离子水清洗柱子,然后填充20%乙醇。拧上柱子上下的堵头,防止柱子变干。
| 货号 | 品名 | 规格 | 用途 | |
| 预装柱 | 29219757 | Superdex 30 Increase 10/300 GL | 10 x 300 mm | 凝胶过滤0-7KDa |
| 29148721 | Superdex 75 Increase 10/300 GL | 10 x 300 mm | 凝胶过滤 3-70 KDa | |
| 28990944 | Superdex 200 Increase 10/300 GL | 10 x 300 mm | 凝胶过滤 10 -600 KDa | |
| 28990945 | Superdex 200 Increase 5/150 GL | 5 x 150 mm | 凝胶过滤 10 -600 KDa | |
| 29091596 | Superose 6 Increase 10/300 GL | 10 x 300 mm | 凝胶过滤 5-5000KDa | |
| 28989333 | HiLoad 16/600 Superdex 75 pg | 16 x 600 mm | 凝胶过滤 3-70 KDa | |
| 28989334 | HiLoad 26/600 Superdex 75 pg | 26 x 600 mm | 凝胶过滤 3-70 KDa | |
| 28989335 | HiLoad 16/600 Superdex 200 pg | 16 x 600 mm | 凝胶过滤 10 -600 KDa | |
| 28989336 | HiLoad 26/600 Superdex 200 pg | 26 x 600 mm | 凝胶过滤 10 -600 KDa | |
| 17090501 | SUPERDEX 30 PREP GRADE 150 ML | 150ml | 凝胶过滤0-7KDa | |
| 17104401 | SUPERDEX 75 PREP GRADE 150 ML | 150ml | 凝胶过滤 3-70 KDa | |
| 17104301 | SUPERDEX 200 PREP GRADE 150 ML | 150ml | 凝胶过滤 10 -600 KDa | |
| 填料 | 17059901 | Sephacryl S-300 HR | 750ml | Sephacryl 凝胶填料 |
| 17058410 | Sephacryl S-200 HR | 150ml | Sephacryl 凝胶填料 | |
| 17003301 | Sephadex G-25 Medium | 100 g | Sephacryl 凝胶填料 | |
| 17003302 | Sephadex G-25 Medium | 500 g | Sephacryl 凝胶填料 | |
| 17004301 | Sephadex G-50 Medium | 100 g | Sephacryl 凝胶填料 | |
| 分子筛Marker | 17036001 | Blue Dextran 2000 10g | 10g | 确定凝胶柱中的空隙体积 |
17、跑胶鉴定
纯化后的蛋白大小和纯度鉴定可用SDS-PAGE跑胶进行鉴定。
17.1 配胶:拿出电泳仪的制胶架,干净的玻璃板,夹紧玻璃板。先配制下层的分离胶。
用烧杯或者锥形瓶按照以下配方进行配制。

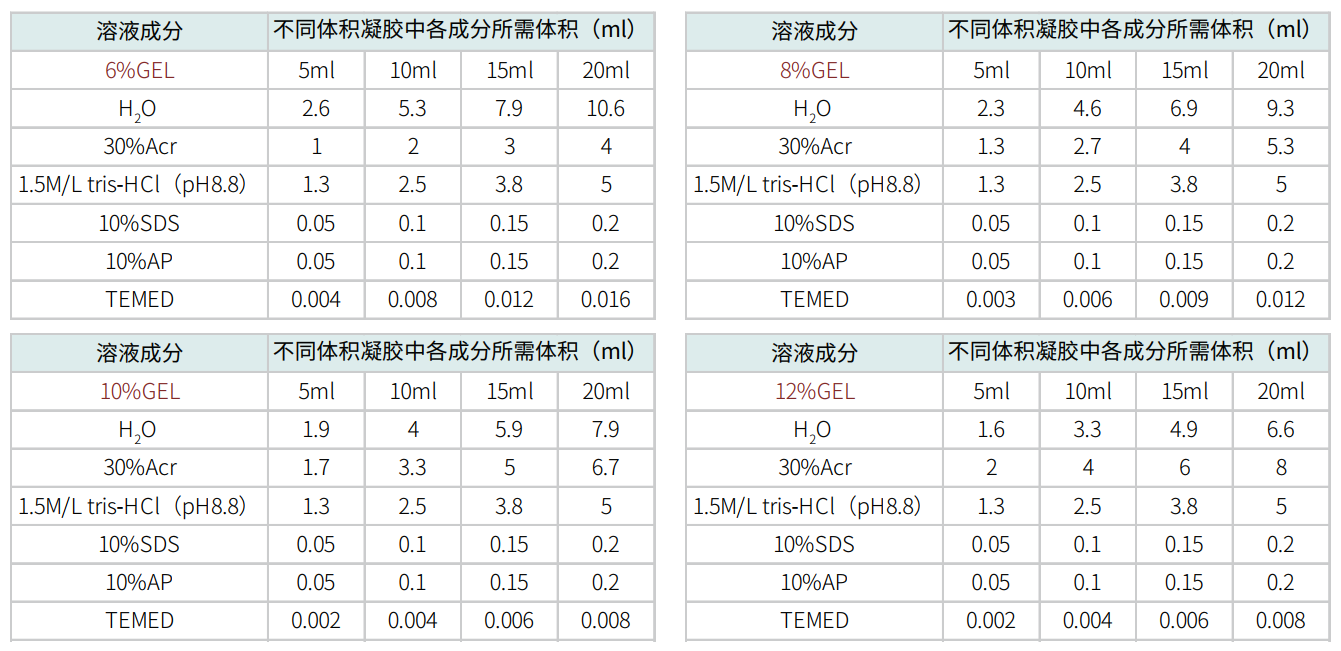

摇匀,用移液枪缓慢加入玻璃板中,注意尽量不要产生气泡。用1ml无水乙醇或者蒸馏水,压在分离胶上面。待分离胶凝固后,倒出上层的无水乙醇或者水,用滤纸吸干。按照以下配方配制上层浓缩胶。
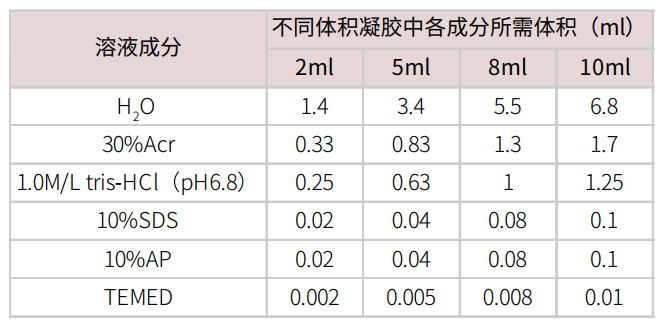
摇匀,用移液枪缓慢加入玻璃板中,注意尽量不要产生气泡。插入梳子。
17.2 上样
把制备好的凝胶从制胶架上取下来,放入电泳槽中。电泳槽加入电泳缓冲液。取部分样品和6xloading buffer混合,在95℃加热5min。拔出胶上的梳子,把样品加到梳孔中。在旁边的孔中加上蛋白Marker。
电泳缓冲液配方:

17.3 跑胶
盖上电泳仪盖子,插上电源。可以先用80V跑胶,当样品跑入分离胶后,可用220V跑胶。溴酚蓝跑到比较靠下的位置,停止跑胶。
17.4 染色
可用考马斯亮蓝R250进行凝胶染色。这里以爱必信的考马斯亮蓝R250产品说明进行介绍。
(1)电泳结束后,取凝胶放入适量考马斯亮蓝染色液中,确保染色液可以充分覆盖凝胶。
(2)置于水平摇床或侧摆摇床上缓慢摇动,室温染色 1h 或更长时间。
注:具体的染色时间取决于凝胶的厚度和染色时的温度。凝胶较厚,温度较低,则染色时间宜适当延长。凝胶较薄,温度较高,则染色时间可以适当缩短。通常染色至凝胶的颜色和染色液的颜色非常接近,在染色液中几乎看不清凝胶时,可以认为已染色充分。
(3)倒出染色液。染色液可以回收重复使用至少 2-3 次。
(4)加入适量考马斯亮蓝染色脱色液,确保脱色液可以充分覆盖凝胶。
(5)置于水平摇床或侧摆摇床上缓慢摇动,室温脱色 4-24h。期间更换脱色液 2-4 次,直至蓝色背景基本上全部被脱去,并且蛋白条带染色效果达到预期。通常蛋白条带在脱色 1-2h 后即可出现。
注:脱色时间过长也会导致蛋白条带的颜色变浅。
(6)完成脱色后,进行观察和拍照。
部分相关产品
| 货号 | 名称 | 规格 | |
| 配胶产品和预制胶 | abs9389 | 预制胶 8%, 10%, 12%, 4-20, 8-20%/10 wells, 15 wells | 10片 |
| abs9367 | 彩色PAGE快速凝胶试剂盒(6%, 8%, 10%, 12%, 15%) | 1Kit | |
| abs9238 | 10%SDS十二烷基硫酸钠溶液 | 500ml | |
| abs42086053 | Sodium Dodecyl Sulfate | 100g/500g/5kg | |
| abs9195 | 1M Tris-HCl,pH6.8 | 1L | |
| abs9201 | 1M Tris-HCl,pH8.8 | 1L | |
| abs9202 | 1.5M Tris-HCl,pH8.8 | 1L | |
| abs9163 | 30%丙烯酰胺/甲叉双丙烯酰胺,19:1 | 500ml | |
| abs9164 | 30%丙烯酰胺/甲叉双丙烯酰胺,29:1 | 500ml | |
| abs9165 | 30%丙烯酰胺/甲叉双丙烯酰胺,37.5:1 | 500ml | |
| abs9166 | 40%丙烯酰胺/甲叉双丙烯酰胺,19:1 | 500ml | |
| abs9167 | 40%丙烯酰胺/甲叉双丙烯酰胺,29:1 | 500ml | |
| abs9168 | 40%丙烯酰胺/甲叉双丙烯酰胺,37.5:1 | 500ml | |
| abs9136 | 甘氨酸 | 500g/1kg | |
| MP12W10 | 预制胶4-20% ,8-16%,8%,10%,12%,10孔、12孔和15孔 | 10片 | |
| 17131301 | SDS十二烷基硫酸钠 | 500g | |
| 17132301 | 甘氨酸 | 500g | |
| 蛋白marker | RPN800E | Amersham ECL Rainbow Marker - Full range | 250ul |
| abs923 | 预染蛋白marker | 500ul/5*500ul | |
| 上样缓冲液 | abs953 | 2*上样缓冲液 | 10ml |



 危险品化学品经营许可证(不带存储) 许可证编号:沪(杨)应急管危经许[2022]202944(QY)
危险品化学品经营许可证(不带存储) 许可证编号:沪(杨)应急管危经许[2022]202944(QY)  营业执照(三证合一)
营业执照(三证合一)