

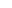



“核小体”是染色体的基本结构单位,一个核小体由两个组蛋白H2A,两个组蛋白H2B,两个组蛋白H3,两个组蛋白H4组成的八聚体和147bp缠绕在外面的DNA组成。
组成核小体核心区域的组蛋白游离在外的N-末端的氨基酸,在相关酶的作用下可以发生不同的修饰。许多组蛋白修饰都能够影响基因的转录活性。
组蛋白的主要修饰类型有:乙酰化、甲基化、磷酸化、泛素化、SUMO化和ADP-核糖化,其中最常见的修饰形式是组蛋白甲基化和乙酰化。组蛋白甲基化和乙酰化状态是否正常通常都会影响各类疾病发生,特别是在肿瘤、免疫等疾病中。
组蛋白甲基化:通过组蛋白甲基转移酶(HMT)将甲基转移至组蛋白突出的“尾巴”中,尤其是N末端的尾巴。组蛋白甲基化通常可发生在精氨酸(R)与赖氨酸(K)残基上,赖氨酸残基可被单甲基化、二甲基化或三甲基化;精氨酸残基则可被单、双甲基化。
组蛋白甲基化修饰既与基因的转录抑制相关,又与转录激活相关,这取决于修饰的不同位点。如转录激活相关的H3K4、K36、K79;转录沉默相关的H3K9、K27、H4K20。
组蛋白乙酰化:通过组蛋白乙酰化转移酶(HAT)将乙酰辅酶A的乙酰基转移到组蛋白氨基末端特定的赖氨酸残基上。组蛋白乙酰化主要通过中和赖氨酸的正电荷从而增加组蛋白末端的黏性,从而促进核小体变得松散,使转录得以发生。所以通常组蛋白乙酰化与转录激活相关,去乙酰则与转录抑制相关。
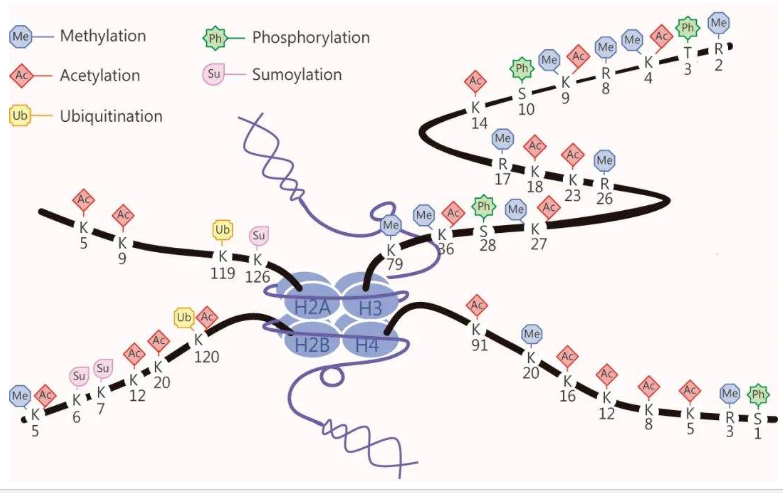
近年来,表观遗传一直是国自然的热点领域,在2019年的21个国自然研究热点中,表观遗传相关热点就占7个。其中,组蛋白修饰作为基因转录层次调控中重要的一环,已在2019年中标项目中超过80项。而且,组蛋白相关文章的发表数在近几年更是热度未减。
因此,组蛋白修饰作为近几年科学研究热点之一,一定有好多老师特别关注关于组蛋白修饰在文章中的研究思路!今天,小优也找了两篇分别关于组蛋白甲基化和乙酰化的高分文章,那我们一起来解读一下他们的研究思路吧!

1
Science: Dual Regulation of Histone Methylation by mTOR Complexes Controls Glioblastoma Tumor Cell Growth via EZH2 and SAM
胶质母细胞瘤(GBM)现在仍然是最常见和致命的人类原发性脑肿瘤,开发针对该疾病的疗法依然需要很大程度的努力。
之前研究表明,在包括胶质母细胞瘤在内的癌症中,H3K27me3(H3K27的三甲基化)水平和癌细胞的生存密切相关,这表明H3K27me3是潜在的诊断标志物和干预治疗靶点。此外,一系列研究表明,药理学干扰H3K27甲基化是治疗脑胶质瘤有效的治疗策略。
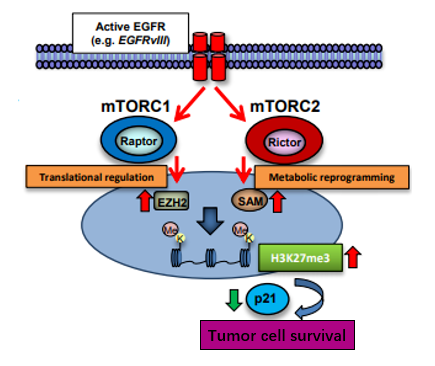
本研究发现H3K27甲基化可能由mTOR1复合物和mTOR2复合物两条通路途径双重调控,并对其机制进行研究。
最后发现mTOR1复合物和mTOR2复合物的共同抑制剂PP242,可以通过阻断以上两条通路途径,进而抑制胶质母细胞瘤的生长和增值。mTOR1复合物和mTOR2复合物成为抗胶质母细胞瘤的的潜在靶标。
1、在胶质母细胞瘤中H3K27三甲基化和EGFR的激活状态密切相关

图1. EGFR突变和扩增与GBM中H3K27me3有关
A.在手术切除的GBM样本中, EGFRvⅢ表达和组蛋白甲基化( H3K27me3 )差异表达的代表图像;
B. 有和无EGFR扩增的GBM病例的H3K27me3 差异表达的免疫荧光和免疫组化。
激活的EGFR突变体Ⅲ( EGFRvIII )与人GBM组织中H3K27me3的表达密切相关, 而且作者进一步发现EGFR扩增与人类GBM病例H3K27me3表达也呈正相关,提示组蛋白甲基化不一定依赖于突变的具体类型,而是取决于EGFR的激活状态。
那EGFR信号激活和H3K27me3之间的机制是什么呢?
2、在GBM细胞中,两个mTOR 复合物会调控H3K27me3水平
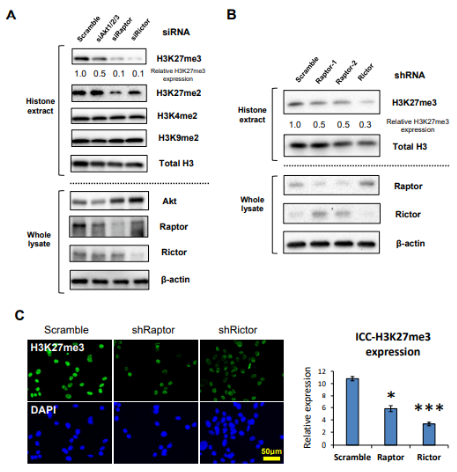
图2. mTOR复合物调控H3K27的三甲基化
A.针对EGFR信号下游效应子Akt、Raptor ( mTORC1 )和Rictor ( mTORC2 )的siRNAs免疫印迹检测U87 - EGFRvIII细胞中H3甲基化标记物( H3K4me2、H3K9me2、H3K27me2、H3K27me3 );
B. 免疫印迹检测转染慢病毒shRaptor ( shRaptor-1,shRaptor-2 )和shRictor的U87 - EGFRvIII细胞H3K27me3变化;
C . Raptor或Rictor的shRNA免疫荧光染色U87 - EGFRvIII细胞H3K27me3 ( 绿色 )。
无论在Raptor ( mTORC1 )和Rictor ( mTORC2 )的siRNAs的U87 - EGFRvIII细胞,还是在染慢病毒shRaptor ( shRaptor-1,shRaptor-2 )和shRictor的U87 - EGFRvIII细胞中H3K27me3水平都显著降低,因此,H3K27me3同时受到EGFR信号异常激活下游mTORC1和mTORC2的调控。
3、mTORC1通过调控H3K27特异性甲基转移酶EZH2的翻译来调控H3K27me3水平

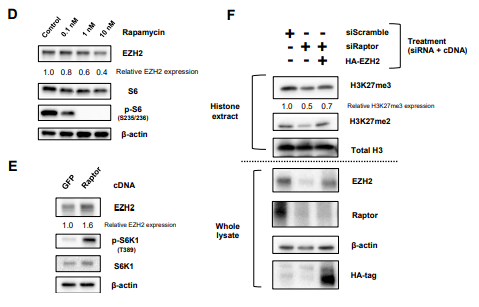
图3. mTORC1调控H3K27特异性甲基转移酶EZH2的翻译
A. 手术切除的EGFRvⅢ- GBM标本,EZH2中差异表达的代表图像;
B .针对EGFR信号中下游分子包括Akt、Raptor ( mTORC1 )和Rictor ( mTORC2 )的siRNA, U87 - EGFRvIII细胞中EZH2的免疫印迹检测;
C. 针对Akt、Raptor或Rictor的siRNA的U87 - EGFRvIII细胞定量免疫荧光染色EZH2 (绿色);
D .免疫印迹评估雷帕霉素处理U87 - EGFRvIII细胞48h后EZH2的变化;
E. 免疫印迹检测EZH2在U87细胞对照( GFP )和过表达Raptor c DNA中的变化。F. H3K27me2和me3在Raptor缺失的U87 - EGFRvIII细胞中和同时过表达后EZH2的免疫印迹检测。
Raptor(mTORC1) 敲除的U87 - EGFRvIII细胞中,EZH2表达受到抑制,但是Rictor ( mTORC2 ) 敲除的U87 - EGFRvIII细胞中,EZH2的表达水平确未受到影响。
用mTORC1的抑制剂雷帕霉素(不同浓度)处理U87 - EGFRvIII细胞48h后,发现EZH2的表达浓度依赖性降低,Raptor cDNA过表达后发现,EZH2增加,另外H3K27me2和me3在Raptor缺失的U87 - EGFRvIII细胞中的减少部分被EZH2的同时过表达恢复。因此,mTORC1是通过组蛋白修饰酶EZH2的翻译调控来促进H3K27me3的表达。
4、mTORC2通过产生S-腺苷蛋氨酸( SAM )促进H3K27的甲基化
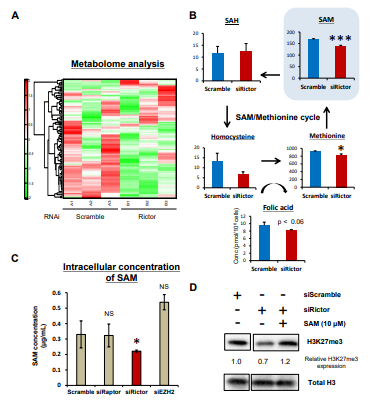
图4. mTORC2调控S-腺苷蛋氨酸( SAM )的生成
A. Rictor的siRNA沉默U87 - EGFRvIII细胞48小时后和空白对照差异表达的代谢物的二维层次聚类的热图;
B. 代谢组学分析显示siRNA沉默Rictor后U87 - EGFRvIII细胞SAM代谢相关代谢产物(蛋氨酸循环); C. 针对不同靶点siRNA沉默后U87-EGFRvIII细胞中SAM的ELISA检测;
D. 免疫印迹法检测转染Rictor siRNA的U87 - EGFRvIII细胞中H3K27me3的变化,并在一定浓度下加入外源SAM作用24小时。
以上结果发现敲除Rictor,并不能改变EZH2的mRNA转录水平和翻译水平,因此推测,mTORC1和mTORC2是通过不同的机制调控H3K27me3水平。
综合代谢组学分析显示mTORC2抑制导致中间代谢产物发生显著变化,其中组蛋白甲基化的底物S-腺苷蛋氨酸( SAM )的胞内浓度在Rictor敲除的U87 - EGFRvIII细胞中显著降低。
值得注意的是,外源SAM的补充恢复了Rictor基因敲除的U87细胞中的H3K27me3水平。因此,mTORC2通过产生组蛋白甲基化底物SAM来调节GBM细胞中的H3K27me3表达。
以上研究表明mTORC1和mTORC2通过不同机制来调控 H3K27me3的表达水平,而mTORC1和mTORC2是否是通过调控H3K27me3的表达水平来促进GBM细胞的增值呢?
5、mTOR复合物通过诱导H3K27me3促进GBM细胞增殖

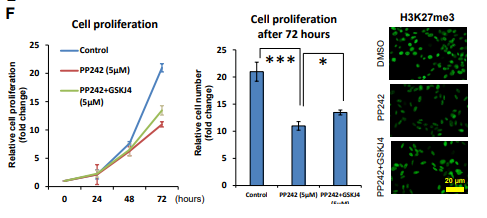
图5. mTOR复合物通过诱导H3K27三甲基化促进
GBM细胞增殖
针对Paptor、Rictor siRNA的以及沉默后用H3K27特异性去甲基化酶的抑制剂GSKJ4处理的U87 - EGFRvIII细胞中H3K27me3的代表性细胞图像( A )和免疫印迹检测(B);
C. GSKJ4作用U87 - EGFRvIII细胞24 h后,非H3K27抑制性组蛋白修饰( H3K9me2和H3K36me2 )无明显变化。
D. 针对Paptor、Rictor siRNA的以及同时用GSKJ4处理2d后的U87 - EGFRvIII细胞增值(细胞数)的差异柱状图;
E. 针对Paptor、Rictor siRNA的以及同时用GSKJ4处理2d后的U87 - EGFRvIII细胞死亡(细胞数)的差异柱状图;
F. PP242 ( 5uM )或PP242 ( 5uM )与GSKJ4 ( 5uM )联合处理U87 - EGFRvIII细胞,在指定时间点测细胞增殖。在相同条件下处理72小时的U87 - EGFRvIII细胞中H3K27me3表达的免疫细胞化学图像。
以上研究表明,两个mTORC通过两个独立、两个相互协调的途径调节与H3K27特定组蛋白甲基化相关的信号级联。
通过对针对Paptor、Rictor siRNA的以及同时用GSKJ4处理GBM细胞,测GBM细胞的增值以及死亡数等,结果表明:mTOR复合物使H3K27me3增加,可加速GBM细胞增殖。
6、本文进一步用两个mTOR复合物的共同抑制剂PP242处理GBM细胞,发现其增值受到很好的抑制
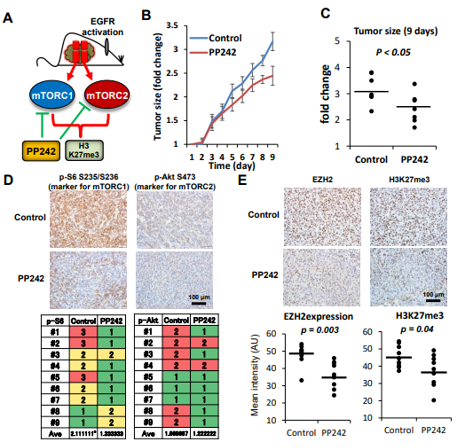

图6. 在GBM ( U87 - EGFRvIII )移植小鼠模型中,mTOR复合物的双重抑制剂伴随着EZH2和H3K27me3表达的降低而抑制肿瘤生长
A. 小鼠异种移植模型示意图;
B . PP242处理的每只移植瘤小鼠在指定时间点皮下肿瘤大小的折线图和散点图(C);
D. 肿瘤组织中活化mTORC1 ( p-S6S235 / S236 )和mTORC2 ( p- AktS473 )标记物在DMSO ( Control )或PP242作用下的定量免疫组化分析。
E . DMSO ( Control )或PP242处理的肿瘤中EZH2和H3K27me3的定量免疫组化分析。
F . DMSO ( Control )或PP242治疗肿瘤p21的定量免疫组化分析;
G. 用DMSO (对照)、PP242或PP242与GSKJ4联合作用8 d的裸鼠移植瘤。
通过建立一个移植的小鼠肿瘤模型,用一种双重mTOR抑制剂( PP242 )进行处理,发现PP242治疗有效地抑制了GBM肿瘤在体内的生长,mTOR标记物、H3K27me3和EZH2表达显著降低。
重要的是,PP242对移植瘤生长的影响部分依赖于H3K27me3的水平,并通过GSKJ4的同时给药证实。这些结果表明mTOR复合物通过诱导H3K27三甲基化促进GBM细胞增殖
本文用到的产品也为大家总结了出来:
2
Cell Metabolism: Nuclear Glycogenolysis Modulates Histone Acetylation in Human
Non-Small Cell Lung Cancers
肺癌是全世界最常见的癌症,其中非小细胞肺癌(NSCLC )约占所有肺癌病例的85%。最近的研究发现,在NSCLC中,异常代谢是转化过程中的重要特征,并且有望成为潜在的新的治疗靶点。
本文通过研究发现malin(E3泛素连接酶)通过泛素化GPBB(糖原磷酸化酶),并促进其核转位,进而调控核糖原分解,增加组蛋白乙酰化,从而减少移植到小鼠体内的癌细胞的增值,阐明了细胞代谢物控制表观遗传调控的一种机制。
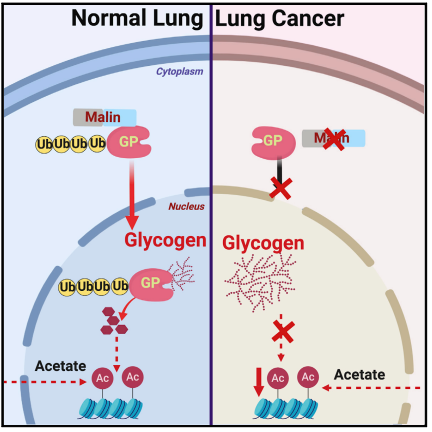
1、糖原合成发生在非小细胞肺癌细胞核内

图1. NSCLC中核糖原累积
A、B. NSCLC肿瘤及邻近良性肺组织(正常)糖原免疫组化染色及HALO数字病理软件定量;
C. NSCLC肿瘤及邻近良性肺组织核糖原的生化定量
NSCLC组织中的糖原含量要远远高于肿瘤远端良性组织中的糖原量,高大约10~100倍,而且良性组织中含有可测量的糖源,表明糖原代谢是一种正常细胞中的生理事件。
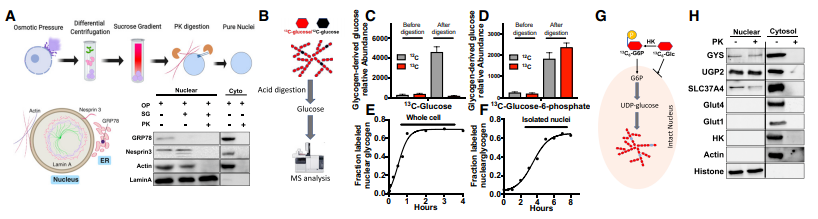
图2. 核内糖原生物合成
A.A549细胞中获得纯核的顺序纯化步骤示意图以及对每个组分进行免疫印迹分析;
B. 糖原标记图式及富集气相色谱-质谱分析;C、D.采用纯化的A549、H1299和H2030细胞完整细胞核,以13C6 -葡萄糖和13C6 -葡萄糖-6 -磷酸为底物进行核糖原合成;
E. 以13C6 -葡萄糖为底物,培养HEK293细胞发生核糖原合成气相色谱-质谱分析结果;
F. 以HEK293细胞纯化的细胞核确定核糖原合成的气相色谱-质谱分析结果;
G. 描述核糖原合成利用13C6 -葡萄糖-6 -磷酸示意图;
H. 采用免疫印迹法检测A549细胞核和细胞质中糖原合成酶( GYS )、UDP-葡萄糖焦磷酸化酶( UGP2 )、己糖激酶( HK )、G6P-转座酶( SLC37A4 )和葡萄糖转运蛋白( Glut1和Glut4 )的含量。
分别用A549的细胞核和全细胞与13C-葡萄糖、13C-葡萄糖-6 -磷酸共同孵育,酸水解后打气相色谱-质谱,发现培养细胞容易利用13C6 -葡萄糖生成核糖原,但是分离的细胞核需要13C6 - G6P作为核糖原合成的底物。
2、核糖原分解依赖于Malin(E3泛素连接酶)调控GPBB(糖原磷酸化酶)的转运
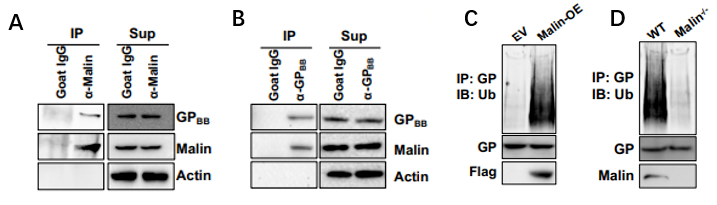
图3. GPBB是Malin的泛素化底物
A、B.GPBB和Malin的Co-IP实验;从WT和小鼠肺组织中分离的A549细胞( C )和AT2细胞( D )中在空载体( EV )和Malin过表达)的细胞系中GPBB泛素化。
通过免疫共沉淀和免疫印迹实验,Malin和GPBB之间有相互作用,而且GPBB是Malin的泛素化底物。
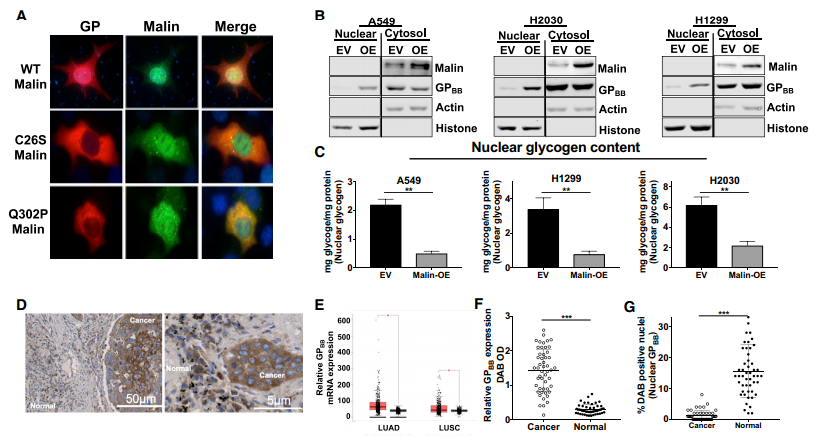
图4. Malin促进GPBB的核定位
A. GP和Malin在野生型或Malin突变体C26S或Q302P共表达的HEK293细胞中的免疫荧光定位;B. 过表达Malin或空载体( EV )的A549、H2030和H1299细胞系GPBB和Malin的细胞核和胞浆组分的免疫印迹分析以及各细胞系中的糖原生化定量(C);D、F. NSCLC组织及正常组织GPBB免疫组化染色和定量分析。G. 定量NSCLC组织和正常样本中GPBB含核百分比。
Malin和GPBB在HEK293细胞中的共表达导致GPBB从细胞质转移到细胞核,而Malin过表达细胞系中的核糖原减少70%;肺癌组织中GPBB大部分分布在胞质中,正常组织中,GPBB大部分分布在胞核中。
突变的Malin样本中,GPBB核转位消失。因此推测我们认为GPBB以Malin依赖的方式转位细胞核,减少的Malin阻碍GPBB向细胞核的转运,导致糖原在细胞核内的积累。
3、核糖原为组蛋白乙酰化提供一个碳源库
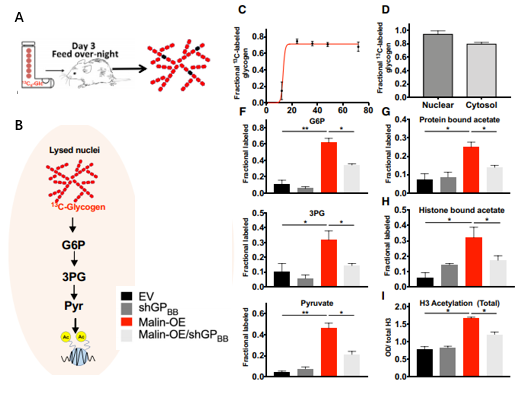
图5. 糖原为细胞核中组蛋白乙酰化提供代谢源
A.13C6 -葡萄糖喂养小鼠产生肝脏13C-糖原示意图;B. 以13C-糖原为底物,用纯化的核裂解液孵育,追踪其代谢过程;
C、D. 13C6葡萄糖液体饲料喂养24h后从小鼠肝脏中分离出胞浆和核糖原的部分标记物。F. 用空载体( EV )、GPBB敲除( shGPBB )、苹果酸过Malin表达和shGPBB对A549、H1299和H2030细胞进行核纯化,裂解液与13C-糖原孵育。用GC-MS对各裂解液生成的13C富葡萄糖-6 -磷酸( G6P )、3 -磷酸甘油酯( 3PG )、丙酮酸进行定量;
G、H、I. 按C中处理的样本中组蛋白结合的乙酸酯、组蛋白结合的乙酸酯和组蛋白乙酰化测定。
葡萄糖-6 -磷酸( G6P )、3 -磷酸甘油酯( 3PG )、丙酮酸都是糖原代谢的中间产物,丙酮酸可提供细胞内的一大部分乙酸盐,乙酸盐是组蛋白乙酰化所必需的。
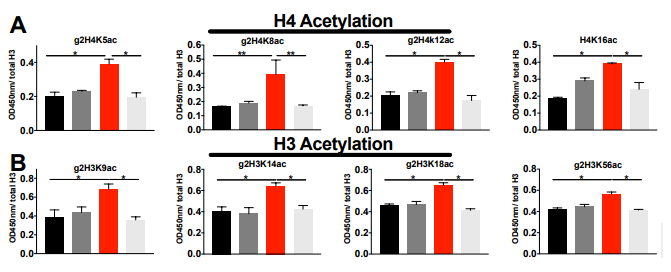
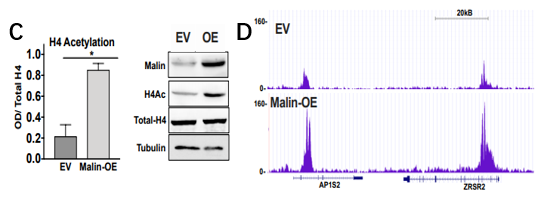
图6. 体外肺癌细胞组蛋白乙酰化及Malin过表达后表观遗传学改变
A、B. 对以上图5C中处理样本的乙酰化H4赖氨酸残基和H3赖氨酸残基ELISA;C. 组蛋白H4乙酰化的ELISA和免疫印迹定量检测;D. EV和Malin过表达中H4Ac的ChIP-seq结果图。
过表达Malin的A549细胞表现为核糖原裂解组蛋白乙酰化增加,直接导致表观遗传学改变,200个基因/位点在Malin过表达后发生变化,其中198个基因/位点上调。因此,Malin可促进糖原分解,促进丙酮酸的产生以及组蛋白乙酰化。
NSCLC组织中的糖原含量要远远高于肿瘤远端良性组织中的糖原,Malin促进糖原分解是否可以抑制NSCLC的生长呢?
4、Malin(E3泛素连接酶)的加入抑制了肺癌细胞在体内的生长

图7. 在体内Malin调节核糖原代谢和组蛋白乙酰化
A.EV 和Malin过表达的指示异种移植瘤生长;B、C、D、E、F 肿瘤移植瘤提取物中Malin的相对丰度(DAB) 、GPBB、糖原、组蛋白H3和H4的乙酰化含量。
在3种非小细胞肺癌细胞系中Malin过表达促进了GPBB的核转位,促进核糖原的分解,增加了组蛋白H3和H4的乙酰化,显着降低了肿瘤的生长,因此增加Malin的表达丰度,会抑制非小细胞肺癌细胞的生长。
本研究确定了一个分子机制,并发现了核糖原的作用超越了简单的能量缓存,成为乙酰化表观遗传转录调控的底物来源。并将细胞代谢和表观遗传建立了联系,为非小细胞肺癌的治疗提供了又一潜在途径。
小优为大家总结了本文研究用到的产品:
大家别忘了,CST还有方便大家研究信号通路初筛,性价比超高的Sampler Kit哦!小优也整理了组蛋白甲基化和乙酰化相关的Sampler Kit 产品,每支抗体平均700元左右,超值、超划算!

每个Sampler Kit除了一抗靶点外,还有对应的二抗哦!
劲爆消息

自2021年1月1日起,CST所有产品都将实施新的价格体系,悄悄告诉您,Sampler Kit产品线涨幅大约在六、七百元左右。所以有需要的老师赶紧上车啦,小优随时在线哦!

