自噬是一种溶酶体降解途径,它不仅有助于提供营养物质,还能够清除有害物质,如错误折叠的蛋白质和入侵的微生物,帮助支持细胞稳态和生存。因此,自噬缺陷与多种人类疾病有关。
在神经退行性病变中,神经元自噬功能紊乱和异常蛋白质聚集折叠是神经退行性疾病的主要病理改变。
Part 1认识自噬与神经退行性疾病
自噬的一些基础知识,小优之前已给大家做过整理,需要的小伙伴可以戳这里了解。
经典的神经退行性疾病有一些关键性蛋白,例如,阿尔茨海默病(AD)的淀粉样前体蛋白(APP)的Aβ和c末端片段(CTF),帕金森病(PD)的突变α-synuclein,亨廷顿病(HD)的聚谷氨酰胺(polyQ)扩增型亨廷顿蛋白(mHtt)。另外,自噬受体的基因突变,如p62、OPTN、NBR1和ALFY/WDFY3,常与神经退行性疾病有关。
越来越多的证据表明,自噬与神经退行性疾病之间的相互作用,不仅自噬活性的降低是疾病的原因之一,而且疾病相关基因的突变也会在不同阶段抑制自噬。
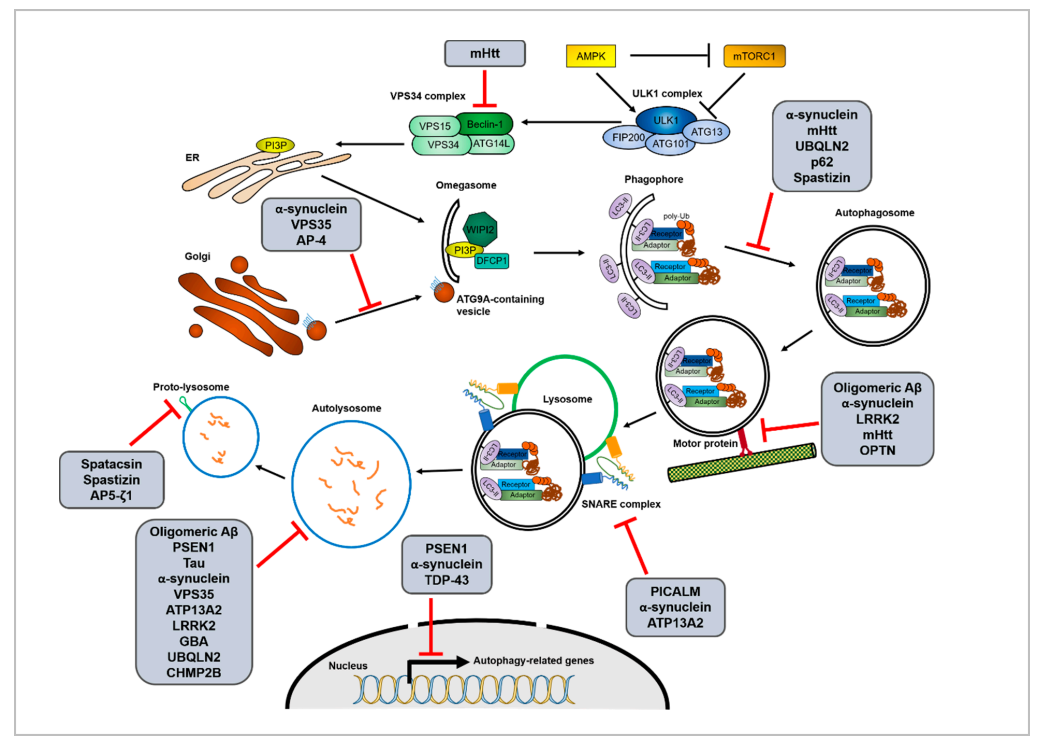
图1:自噬过程与神经退行性疾病相关蛋白之间的相互作用
△点击放大图片
接下来,我们整理了常见的神经退行性疾病(阿尔兹海默病AD、帕金森氏病PD、亨廷顿氏病HD、肌萎缩性侧索硬化症ALS)与自噬机制的前沿进展,以及自噬调节作为这些疾病的治疗策略。
Part 2 自噬途径与神经退行性疾病发病机制的关系
2.1阿尔兹海默病(AD)
阿尔茨海默病(AD)是最常见的痴呆形式,其特征是Tau和β-淀粉样肽两种蛋白质的积累。自噬途径的缺失已被证实是AD的标志。
针对自噬与AD的2种特征蛋白的研究已有很多。我们首先来看,将M1C细胞暴露于3-MA (2.5 mM, 5 mM)中,与未处理对照组相比,用3-MA处理细胞可导致Tau蛋白显著积累(图2A)。此外,在加入3-MA (5 mM)的Tau蛋白诱导5天后,从细胞裂解液中检测到p44阳性Tau蛋白(图2B)。
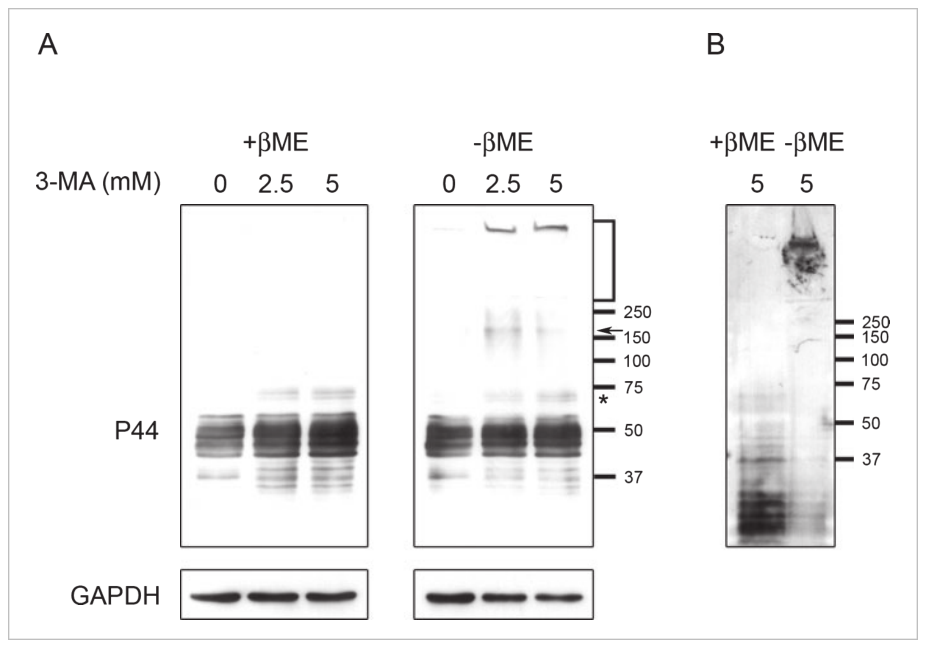
图2:抑制自噬会增加tau蛋白的积累
△点击放大图片
这说明,自噬抑制剂3-MA对细胞的处理会导致Tau蛋白更多的积累,提示自噬系统在清除聚集的Tau蛋白方面发挥着明显的作用[1]。
另一种蛋白淀粉样β肽是淀粉样前体蛋白(APP)降解的结果。淀粉样β肽的积累导致自噬小体与溶酶体融合受损,进而导致蛋白质的积累。此外,编码早衰素1 (PSEN1)的基因的突变也会导致溶酶体功能受损,淀粉样β-肽积累。在AD中淀粉样β-肽的积累和PSEN1突变受到UPR和自噬的保护。只有细胞内淀粉样β-肽和PSEN1突变才能通过RYR和ITPR从内质网释放钙离子来触发内质网应激。有趣的是,PSEN1突变会阻断内质网应激传感器,从而使神经元细胞更容易受到内质网应激的影响。
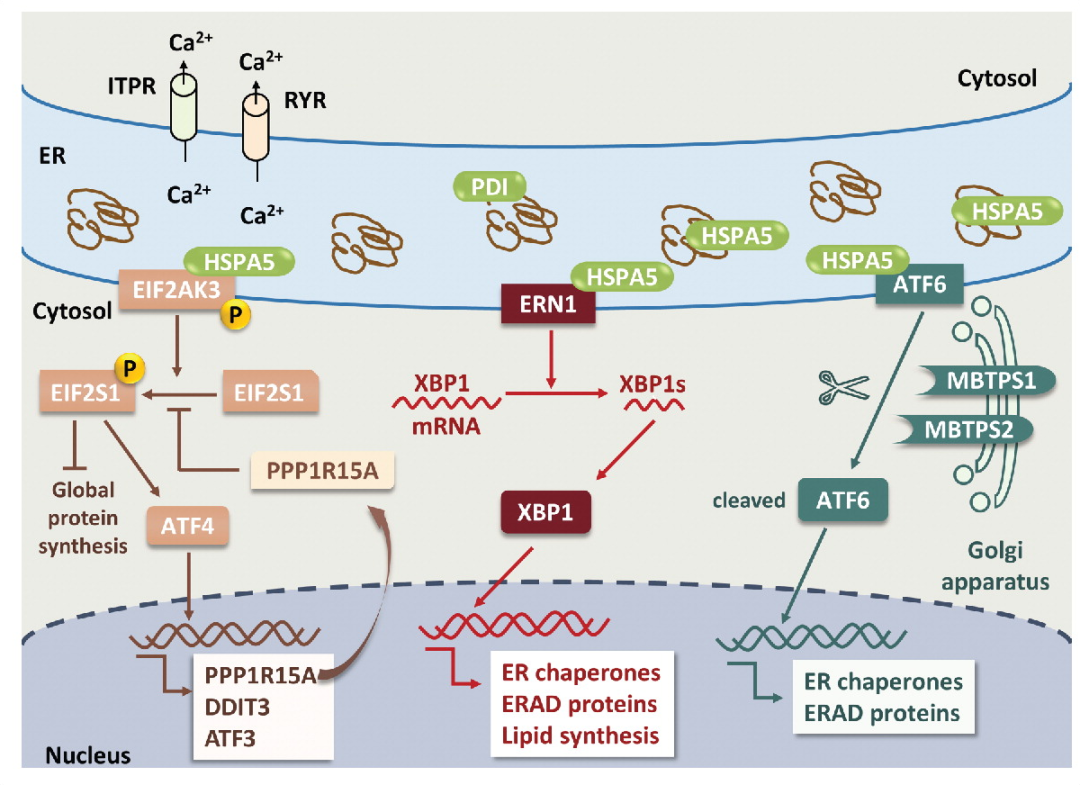
图3:神经元细胞中的 ER 应激和 UPR 通路
△点击放大图片
除内质网应激外,溶酶体pH的改变在AD中也有有效作用。V0 - ATP酶对降低自噬溶酶体的pH值至关重要。已经发现PSEN1突变导致了V0-ATP酶 a1亚基从内质网到溶酶体的靶向性缺陷。因此,PSEN1突变导致AD患者通过自噬减少了底物的蛋白水解,因为PSEN1突变患者的pH没有得到适当的维持[2]。

图4:PS1 KO囊胚中v-ATPase的溶酶体靶向功能受损
△点击放大图片
由于自噬过程中自噬小体酸化和组织蛋白酶激活的选择性损伤,导致基质蛋白水解和自噬小体清除被阻止。引起早发性AD的PS1突变在AD患者的成纤维细胞中产生类似的溶酶体/自噬表型。因此,PS1对于v-ATPase靶向溶酶体、溶酶体酸化和自噬过程中的蛋白水解至关重要。
2.2 帕金森氏病(PD)
帕金森病是一种进行性神经退行性运动障碍,特征是黑质多巴胺能神经元的丢失,以及多巴胺的丢失导致运动障碍。其主要病理特征包括α-synuclein等基因的遗传突变。帕金森病已被检测到包含6个基因的突变,即SNCA、GBA、Parkin (PARK2)、PTEN诱导的推定激酶1 (PINK1)、富含亮氨酸重复激酶2 (LRRK2)和DJ1,这些基因与疾病的早期发生有关。
自噬相关基因的转录改变在PD中很常见。在PD症状期,中脑大部分多巴胺能神经元已经缺失,TFEB介导的Beclin-1、CTSD和LAMP1的转录较症状前期降低[3]。
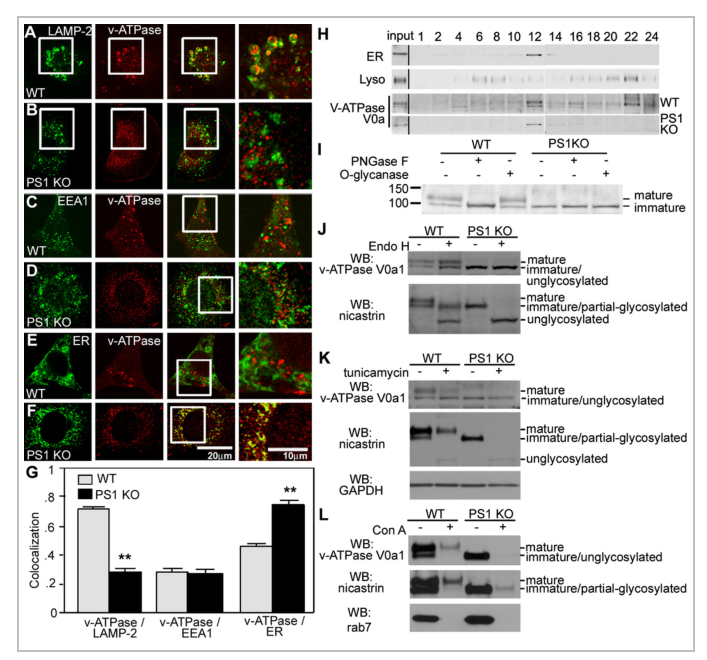
图5:基因转移介导的自噬刺激保护DA神经元免受α-syn毒性。
△点击放大图片
研究显示,过表达Beclin-1和TFEB对黑质DA神经元具有强大的神经保护作用,可抵抗α- sync诱导的毒性。测量结果显示,与GFP对照组相比,Beclin-1和TFEB过表达组纹状体TH+末端存活显著。
葡糖脑苷脂酶(GBA)的突变,是一种降解葡糖神经酰胺的溶酶体酶,是帕金森病最常见的遗传危险因素。研究发现,散发性帕金森病患者在早期表现为GBA活性选择性降低,伴有α-突触核蛋白内含物增加[4]。
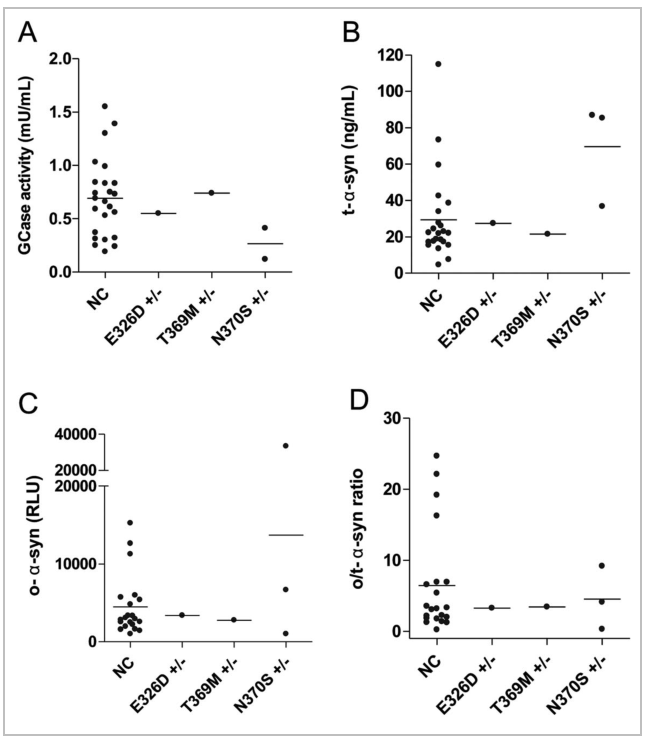
图6:GBA1突变PD患者和nc患者的GCase活性和α-syn物种水平
△点击放大图片
在N370S GBA1突变携带者中,我们观察到比非携带者更低的GCase活性,而这些病例也显示出比非GBA1突变PD患者更高的t-α-syn水平。
帕金森病中第二常见的遗传危险因素是富含亮氨酸重复激酶2(LRRK2/PARK8)基因突变。然而,LRRK2在自噬中的作用是否与PD病理相关仍存在争议。有研究表明,LRRK2缺失会破坏自噬-溶酶体途径,导致细胞死亡[5]。
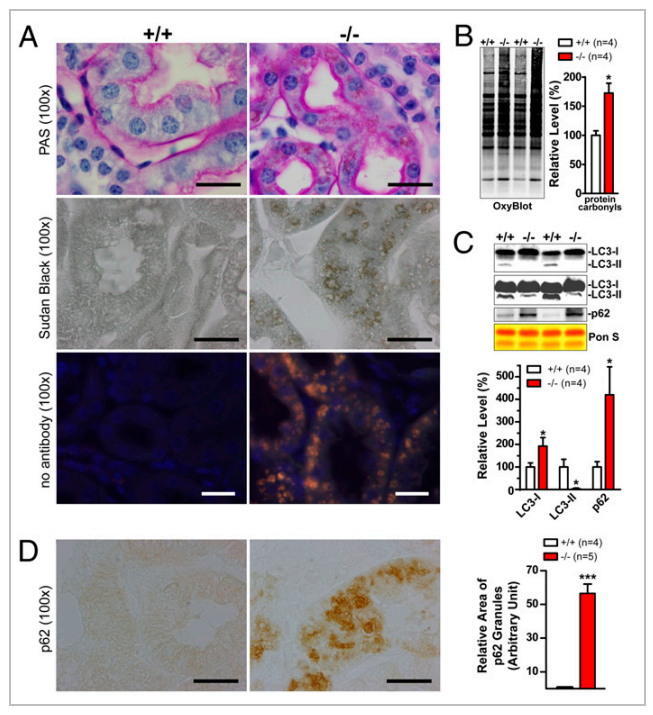
图7:衰老LRRK2−/−小鼠的自噬-溶酶体途径受损,氧化损伤增加
△点击放大图片
图7A,年老的LRRK2−/−小鼠肾脏中脂褐素颗粒异常堆积,脂褐素颗粒的异常积累提示自噬-溶酶体系统受损,并与神经退行性疾病(如PD)有关。为了进一步评估LRRK2缺失情况下的自噬功能,作者还评估了自噬小体的标记物LC3。WB分析显示,20月龄LRRK2−/−肾脏中LC3-II水平显著下降(图7C),表明LRRK2缺失时自噬小体形成受损。
ATP酶离子转运的功能缺失突变体ATP13A2是PD早期发病的特征,该蛋白主要是利用水解三磷酸腺苷释能驱动物质跨膜运输,因此对于维持溶酶体的pH至关重要。研究表明,ATP13A2的突变被证明可以引起α-synuclein的积累,而α-synuclein的沉默可以减轻ATP13A2缺失引起的神经毒性[6],这表明ATP13A2的缺失可能通过α-synuclein的积累参与PD的病理过程。
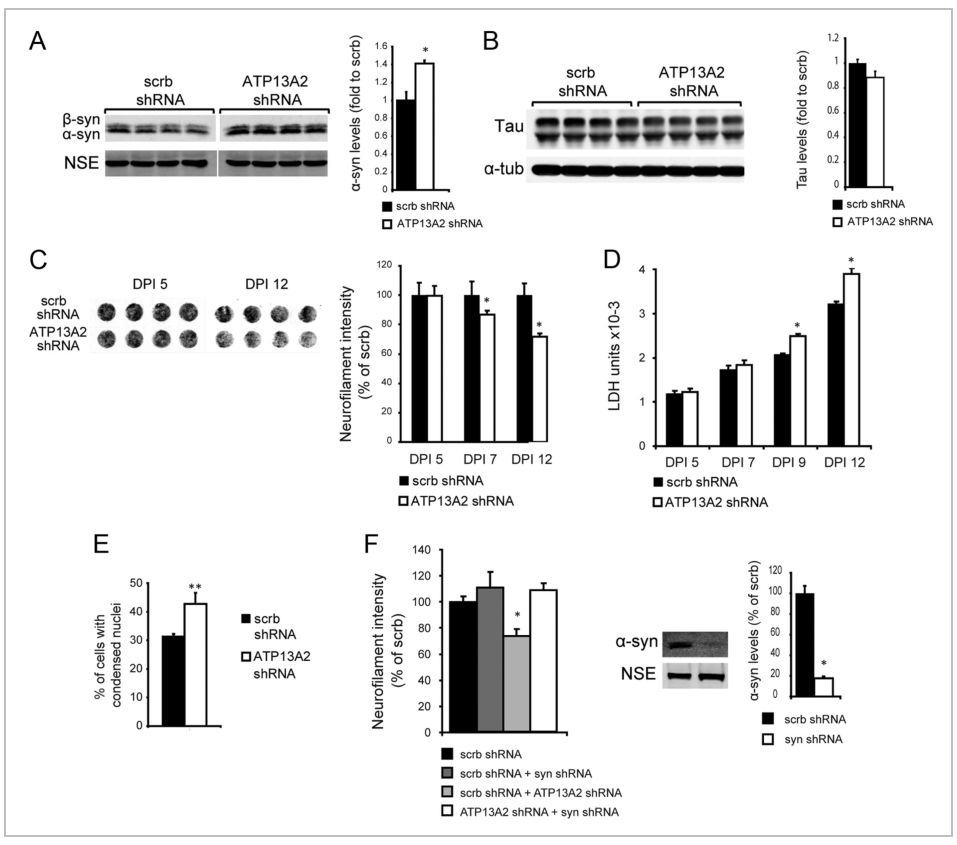
图8:ATP13A2的消耗导致α-syn和初级神经元的毒性
△点击放大图片
这些实验表明,在神经元中敲低ATP13A2导致NF染色减少(图8C), LDH释放增加(图8D),核凝结增加(图8E),提示ATP13A2功能的丧失导致神经元毒性。
2.3亨廷顿氏病(HD)
亨廷顿氏病HD是一种常染色体单基因显性遗传性神经退行性疾病,它会导致主要是身体状态,思维认知能力和情绪的变化。亨廷顿病患者体内的异常亨廷顿蛋白首先会影响其脑内的基底核,使得基底核无法修饰或抑制大脑的指令,于是全身肌肉便不受控制地运动,表现为舞蹈样动作。到了疾病的晚期,连负责下达指令的大脑表层也会逐渐死亡,届时病人可能失去所有行动能力,并出现认知功能下降甚至痴呆。导致亨廷顿氏病的蛋白是突变型的亨廷顿蛋白mHtt,这些蛋白形成泛素阳性聚集物,具有丰富的β-sheet结构,导致纹状体和皮层的细胞毒性。
研究表明,mHtt的过表达可诱导进行性运动障碍,并伴有自噬小体的积累。
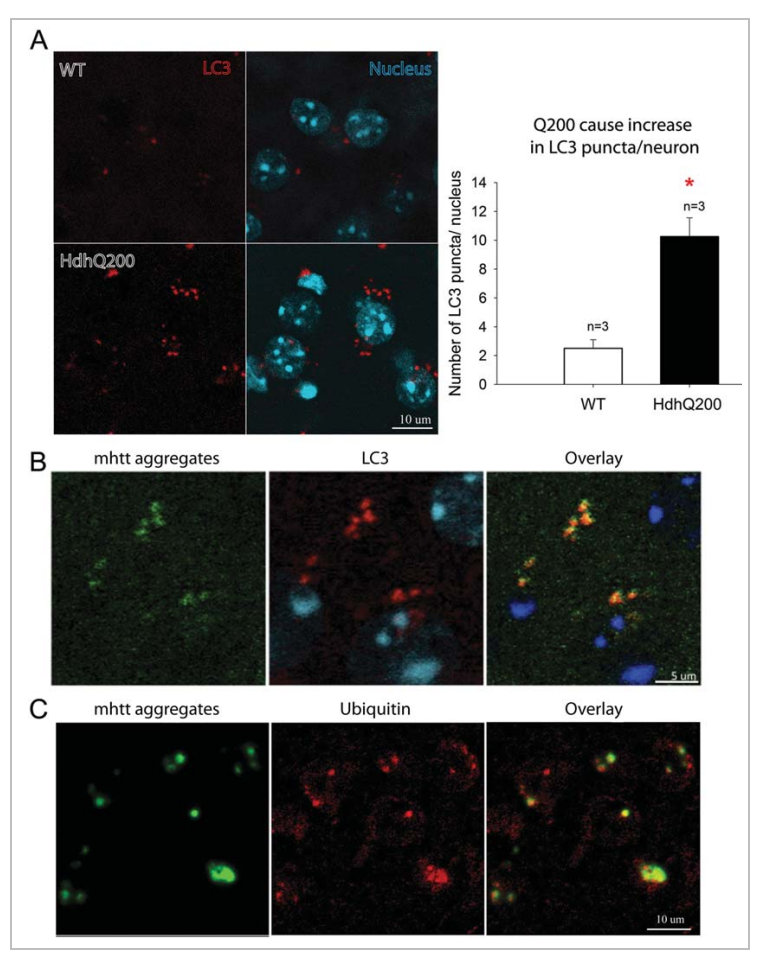
图9:LC3点状蛋白和泛素与mhtt聚集物共定位
△点击放大图片
40周龄HdhQ200杂合子小鼠纹状体相邻部分的LC3免疫荧光标记显示,与WT同窝小鼠对照相比,每个神经元的LC3点状点增加了4倍(图9A)。LC3与AF共定位(图9B),一些异常的蛋白聚集物通过泛素化作用被导向自噬-溶酶体降解途径。
最近对纹状体的全基因组筛查表明,许多自噬相关基因,如Atg4b、TFEB和Atlastin 3,似乎可以预防mHtt毒性。
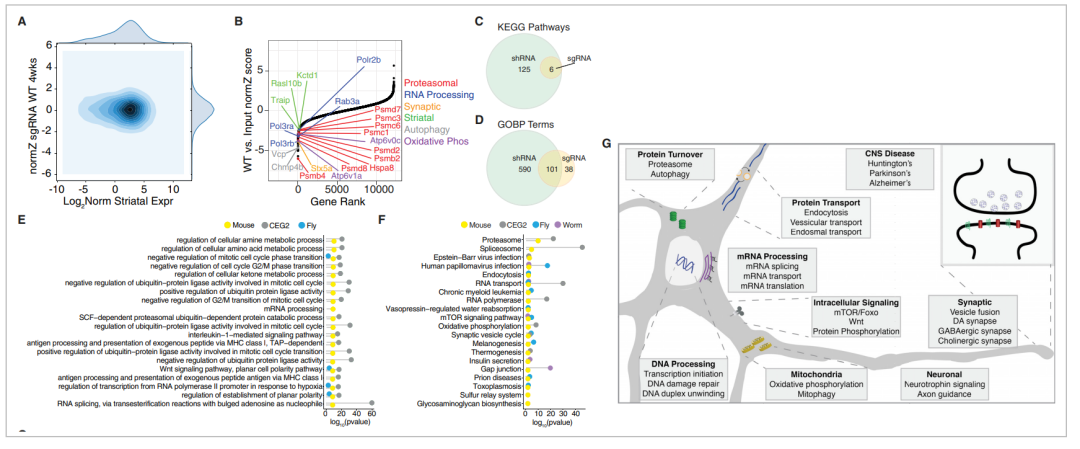
图10:体内CRISPR筛选验证神经元必需基因和通路
△点击放大图片
全基因组筛选已经发现了在成人中枢神经系统中参与维持体内神经元活力的基因。这些基因属于许多细胞通路,包括自噬通路、神经营养蛋白信号和突触传递相关的通路(图10G)。研究人员还在这些重要基因中发现了一组纹状体富集基因,并预测抑制这些特定的纹状体富集基因的mHTT诱导的转录失调将有助于抑制mHTT毒性。
2.4肌萎缩性侧索硬化症(ALS)
肌萎缩性侧索硬化症是一种衰弱性神经肌肉疾病,以脊髓和大脑运动神经元的进行性退化为特征。这些运动神经元的退化导致神经肌肉失神经、随意骨骼肌萎缩,最终导致瘫痪和死亡。目前还没有治愈这种致命疾病的方法,现有的治疗方法效果都比较有限。
迄今为止发现的几个ALS相关基因都与自噬和线粒体吞噬功能有关,特别是蛋白质聚集物和受损线粒体的清除。已知在自噬中起作用的ALS基因包括OPTN、TBK1和SQSTM1等。
这其中,TDP-43是一个多功能的DNA和RNA结合蛋白,在细胞内的RNA转录、选择性剪接及mRNA稳定性调节等过程中发挥功能。在ALS和额颞叶变性(FTLD)病人脊髓或大脑受损区域的神经元和胶质细胞中,能检测到泛素化的蛋白质包涵体,TDP-43是其特征性成分。研究表明,自噬激活能减少TDP-43的聚集,并可改善携带TDP-43突变的人类运动神经元的生存[7]。
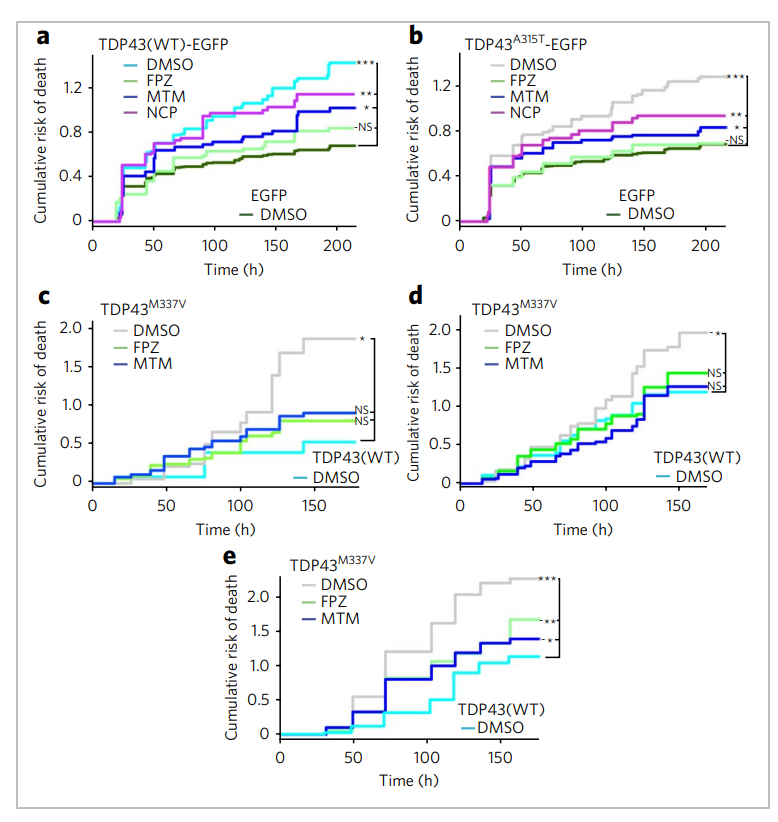
图11:自噬刺激改善ALS神经元和星形胶质细胞模型的生存。
△点击放大图片
研究发现,携带ALS相关TARDBP突变的人iPSC衍生的神经元和星形胶质细胞会积累突变的TDP-43蛋白,这进一步支持了过量的TDP-43是疾病发病机制的中心的观点。并且,自噬诱导有效地提高了啮齿动物神经元和人类iPSC来源的神经元和星形胶质细胞的存活率。这一研究表明增强TDP-43清除是一种合理的人类疾病的神经保护策略。
UBQLN2是另一个家族性ALS的遗传风险因子,也与自噬有关。UBQLN2与LC3形成复合物,并在自噬小体中泛素化货运蛋白。并且,UBQLN2会促进自噬体-溶酶体融合[8]。
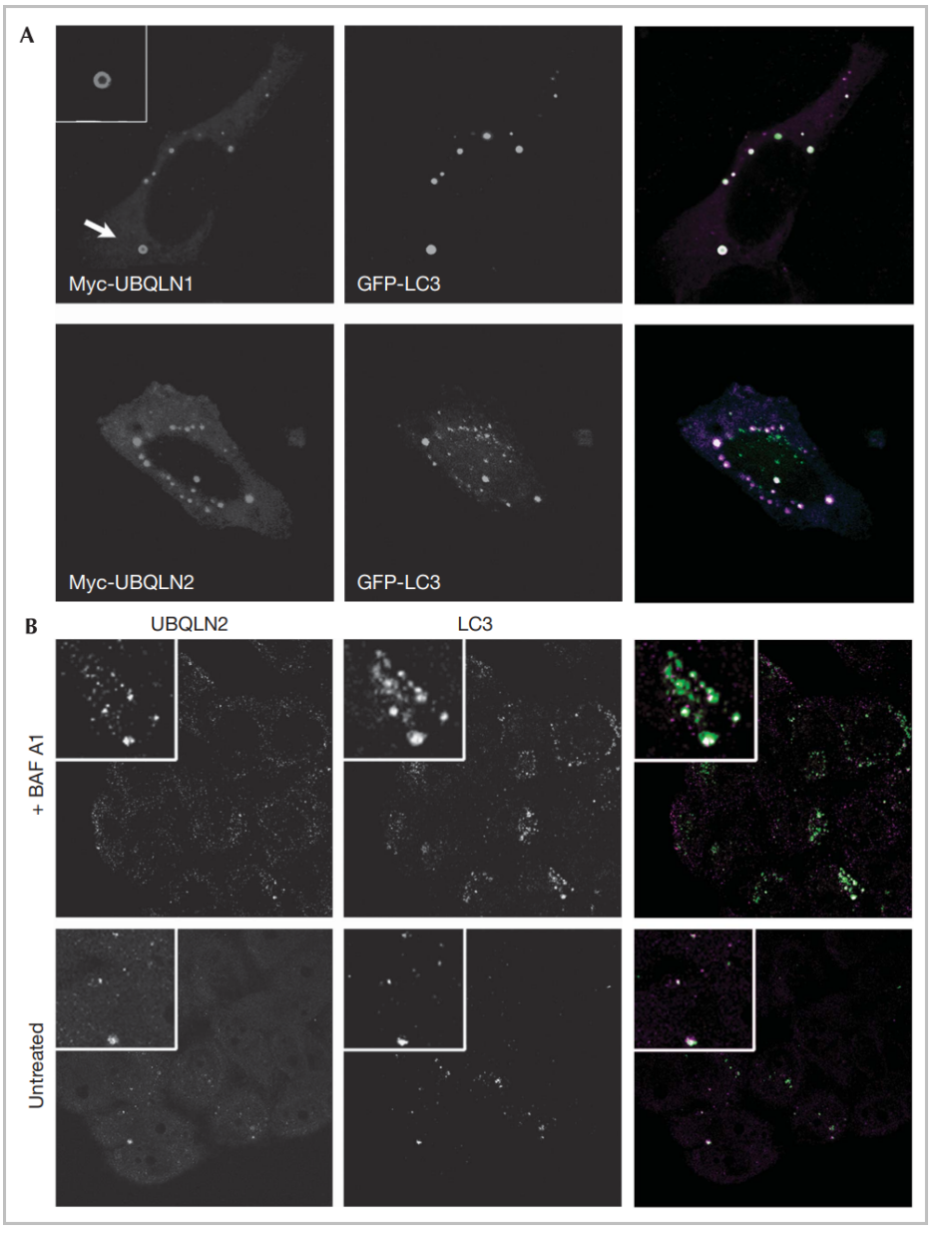
图12:泛素与自噬小体共定位,揭示UBQLNs与自噬小体相互作用
△点击放大图片
结果显示,GFP-LC3广泛共定位,这是自噬小体的标记物(图12A)。UBQLNs和LC3也在内源性稳定表达GFP-LC3的MCF-10A细胞中共定位(图12B),这排除了过度表达的假象。
近年来,ALS与p62、OPTN等聚集性受体蛋白的相关性越来越明显。我们要知道,大约一半的ALS相关突变是位于p62的PB1、LIR和UBA区域,这些区域负责与货运蛋白或LC3的相互作用,这说明源自p62突变的ALS病理可能与蛋白聚集物向自噬小体传递效率低下有关。研究显示,p62过表达会减弱由不溶性蛋白聚集物引起的ALS症状。
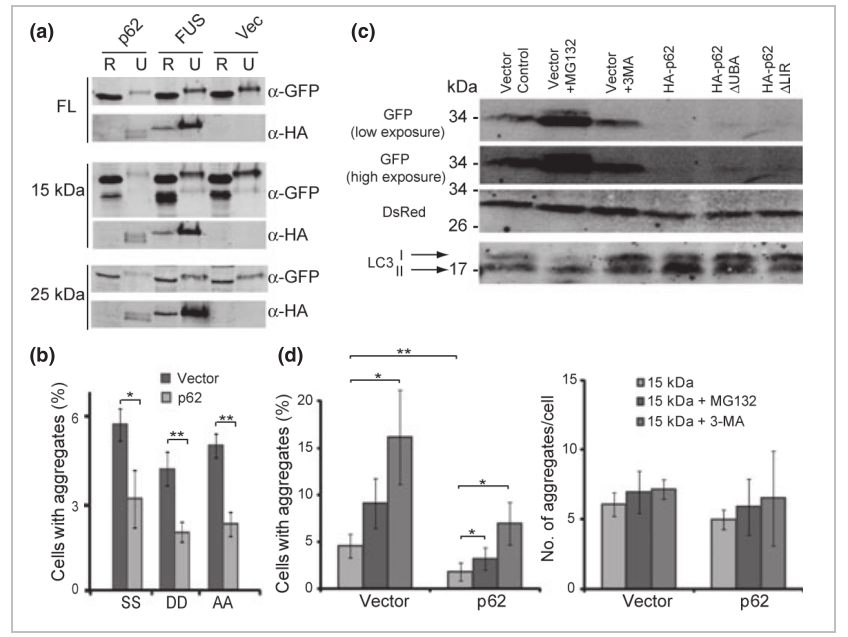
图13:p62的过表达降低了TDP-43的聚集
△点击放大图片
p62在N2A细胞中过表达会导致不溶性TDP-43或TDP-43片段减少(图13a), TDP-43的聚集显著减少(图13b),因此p62水平的增加可以减少TDP-43的聚集。并且在证实了d1GFP蛋白水平可以用作蛋白酶体活性的一个指标后,当p62过表达时,d1GFP信号显著降低,表明p62过表达确实刺激了蛋白酶体的活性(图13c)。作者还以LC3-I向LC3-II的修饰为指标检测了p62对自噬的影响,N2A细胞中p62的过表达导致了更多的LC3-II,这证实了p62的过表达也刺激了自噬(图13c)。
Part 3 自噬上调作为神经退行性疾病的治疗策略
经过我们上面的分析,通过自噬上调来减少细胞内聚集物蛋白的积累,是有助于延缓AD、PD、HD和ALS的疾病恶化的,这表明自噬诱导可作为大多数神经退行性疾病的治疗策略。然而,广泛的自噬对维持细胞内稳态是有害的。因此,用自噬的方法来治疗神经退行性疾病时应谨慎。
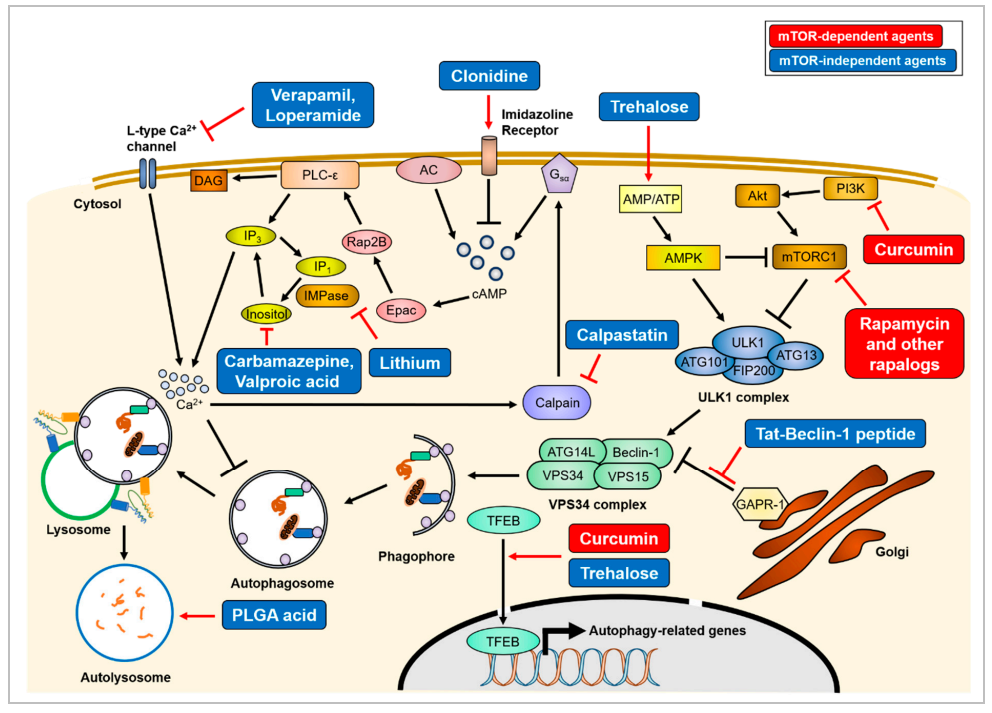
图14:自噬的治疗机制
△点击放大图片
各种自噬诱导剂的处理可以以mTOR依赖性或非依赖性的方式增强聚集倾向蛋白的清除(图14)。mTOR依赖的自噬诱导物(红色),如雷帕霉素和姜黄素,会直接抑制mTORC1的活性,导致ULK1复合物的激活。相反,mTOR非依赖性药物(蓝色)则是通过各种细胞内信号级联或溶酶体生物发生上调自噬活性。
mTOR依赖的自噬诱导剂 作用机制
雷帕霉素 1、抑制mTOR通路,促进自噬发生;2、清除大量有聚集倾向的polyQ蛋白。
姜黄素 提高自噬相关蛋白的表达水平,如Beclin-1、ATG5或ATG16L1,增加自噬通量,清除细胞内聚集物。
mTOR非依赖的自噬诱导剂 作用机制
海藻糖 通过激活AMPK或TFEB来增强自噬,在保护线粒体功能,延缓ALS骨骼肌的退化,对抗凋亡的发生等多个方面发挥神经保护作用。
情绪稳定剂,如维拉帕米、洛哌丁胺、可乐定和钙蛋白酶抑制素等 可通过降低肌醇磷酸3 (IP3)的水平,来诱导自噬-溶酶体系统对聚集性蛋白的降解。
锂盐 抑制肌醇单磷酸酶(IMPase)
卡马西平和丙戊酸 抑制肌醇合成
聚乙醇乳酸 使用聚乙醇乳酸(PLGA)酸性纳米颗粒恢复溶酶体pH值已被证明可以用来修复溶酶体缺陷和自噬降解。
Tat-Beclin-1肽 Tat-Beclin-1小肽是Beclin-1 (aa 267-284)与HIV-1 Tat蛋白结合的细胞渗透性肽。Tat-Beclin-1通过与GAPR-1/GLIPR2相互作用,增强自噬的启动。
部分相关产品:
| 货号 | 产品名称 |
| 12741S | LC3A/B (D3U4C) XP ® Rabbit mAb |
| 86060S | LC3A (E5C9B) Rabbit mAb |
| 83506S | LC3B (E5Q2K) Mouse mAb |
| 14723S | LC3C (D1R8V) Rabbit mAb |
| 9980S | Atg5 (D5G3) Rabbit mAb |
| 4180S | Atg12 (D88H11) Rabbit mAb |
| 8089S | Atg16L1 (D6D5) Rabbit mAb |
| 39749S | SQSTM1/p62 (D1Q5S) Rabbit mAb |
| 16177S | Phospho-SQSTM1/p62 (Ser349) (E7M1A) Rabbit mAb |
| 4122S | Beclin-1 (2A4) Mouse mAb |
| 84966S | Phospho-Beclin-1 (Ser15) (D4B7R) Rabbit mAb |
| 6439S | ULK1 (D9D7) Rabbit mAb |
| 14205S | Phospho-ULK1 (Ser638) (D8K9O) Rabbit mAb |
| 43110T | Mitophagy Antibody Sampler Kit |
| 4445T | Autophagy Antibody Sampler Kit |
| 59187T | Beclin-1 Complex Antibody Sampler Kit |
| 8359T | ULK1 Antibody Sampler Kit |
| abs815906-1mg | Bafilomycin A1 |
| abs817875-5mg | Wortmannin |
| abs810030-10mg | Rapamycin |
| abs810575-50mg | 3-MA |
| abs880011-1kit | Autophagy agonist and inhibitor kit |
参考文献:
[1]Hamano T, Gendron TF, Causevic E, et al. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci. 2008;27(5):1119-1130. doi:10.1111/j.1460-9568.2008.06084.x
[2]Lee JH, Yu WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141(7):1146-1158. doi:10.1016/j.cell.2010.05.008
[3]Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Björklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc Natl Acad Sci U S A. 2013;110(19):E1817-E1826. doi:10.1073/pnas.1305623110
[4]Parnetti L, Chiasserini D, Persichetti E, et al. Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson's disease. Mov Disord. 2014;29(8):1019-1027. doi:10.1002/mds.25772
[5]Tong Y, Yamaguchi H, Giaime E, et al. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci U S A. 2010;107(21):9879-9884. doi:10.1073/pnas.1004676107
[6]Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J Neurosci. 2012;32(12):4240-4246. doi:10.1523/JNEUROSCI.5575-11.2012
[7]Barmada SJ, Serio A, Arjun A, et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol. 2014;10(8):677-685. doi:10.1038/nchembio.1563
[8]N'Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J, Brown EJ. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 2009;10(2):173-179. doi:10.1038/embor.2008.238
[9]Saha S, Panigrahi DP, Patil S, Bhutia SK. Autophagy in health and disease: A comprehensive review. Biomed Pharmacother. 2018;104:485-495. doi:10.1016/j.biopha.2018.05.007
[10]Park H, Kang JH, Lee S. Autophagy in Neurodegenerative Diseases: A Hunter for Aggregates. Int J Mol Sci. 2020;21(9):3369. Published 2020 May 10. doi:10.3390/ijms21093369