蛋白质糖基化是在糖基转移酶的催化下与蛋白质上的某些氨基酸残基形成糖苷键的过程,是生物体内最重要的翻译后修饰形式之一,可发生在细胞中50%-70%的蛋白质上。糖基化在组织和细胞中参与调控蛋白质的定位、功能和活性,会影响细胞识别、分化、信号转导、免疫应答等多种重要的生命活动。
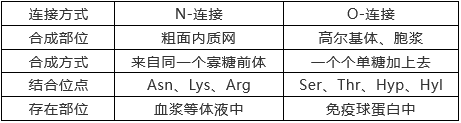
根据糖蛋白糖侧链与蛋白氨基酸基团的不同连接方式,蛋白质糖基化分为不同的修饰类型,其中N-糖基化和O-GlcNAc糖基化是两个主要的修饰类型。N-糖基化和O-GlcNAc糖基化的连接方式、合成部位、氨基酸修饰位点等都不相同。
△点击放大图片
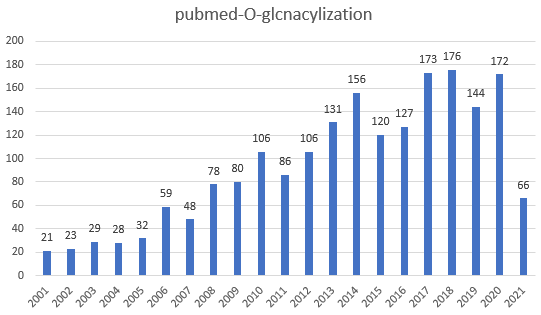
其中O-GlcNAc糖基化没有固定的特征序列,其糖链没有固定的核心结构,因此O-GlcNAc糖基化糖蛋白的结构更加复杂。O-GlcNAc糖基化近几年研究热度更是逐年增加,有研究表明:O-GlcNAc糖基化被认为是一种营养和应激传感器,参加转录和翻译到信号转导和代谢细胞过程的调控。O-GlcNAc糖基化异常可导致肿瘤、糖尿病、心血管和神经退行性疾病等多种病症。
△点击放大图片
小优也精心研读了两篇关于O-GlcNAcylation研究的高分文章,希望能给大家提供一些研究思路啦~
01 Molecular Cell
葡萄糖诱导的O-GlcNAc对脂质从头合成的转录后调控

美国乔治华盛顿大学裴华东与国家蛋白质科学中心(北京)秦伟捷以及贺福初院士团队
△点击放大图片
肿瘤细胞中往往会发生葡萄糖和谷氨酰胺等代谢通路的异常激活,而葡萄糖与谷氨酰胺都是细胞内己糖胺生物合成途径(HBP)的重要底物,该途径是产生糖基供体UDP-GlcNAc的关键通路。糖基转移酶OGT可以利用UDP-GlcNAc在蛋白的丝氨酸或苏氨酸残基上形成O-GlcNAc修饰。O-GlcNAc修饰已被发现参与调控细胞内多种重要的生物学过程,如基因转录和细胞代谢等,但细胞中O-GlcNAc修饰的“reader”还并不清楚。不仅如此,在多种肿瘤组织中,O-GlcNAc修饰的整体水平都有显著上调,因此鉴定其中O-GlcNAc修饰的重要底物与相应的功能机制研究显得尤为关键。
除了葡萄糖代谢异常外,肿瘤细胞还需要其他大量的代谢中间产物来满足其快速生长与增殖的需求,其中就包括脂质成分。尽管肿瘤细胞可以通过从外界微环境中摄取脂质成分,但是研究表明大多数肿瘤细胞中都会发生脂质从头合成途径的异常激活。SREBPs是调控细胞脂质合成的关键转录因子。当细胞内脂质水平下调时,SREBPs蛋白可以发生蛋白切割活化,转位进入细胞核内,从而激活下游脂质合成相关基因的转录与表达。此外,细胞的脂质从头合成也在转录后水平上受到严格的调控。丝氨酸/精氨酸蛋白特异性激酶2(SRPK2),作为重要的mRNA剪接调控蛋白激酶,可以被mTORC1通路的关键下游蛋白激酶S6K1磷酸化修饰,从而增强其细胞核定位,促进脂质从头合成相关基因mRNA的剪接和稳定性,以及肿瘤细胞的脂质从头合成水平与肿瘤生长。因此,SRPK2是阻断脂质生成和抑制mTORC1驱动肿瘤生长的潜在治疗靶点。
本文研究表明UDP-GlcNAc在前体mRNA剪接和脂质从头合成中起着关键作用。OGT-SRPK2通过诱导多个mRNA的有效剪接而不是影响编码基因的转录来促进脂肪酶的产生。O-GlcNAc修饰通过增强SRPK2的细胞核内定位、在转录后水平上调控肿瘤细胞的脂质从头合成水平,进而促进肿瘤生长的新机制;这一调控依赖于importin α/β入核转运系统,进一步研究提示importin α蛋白可能是O-GlcNAc修饰的“reader”。
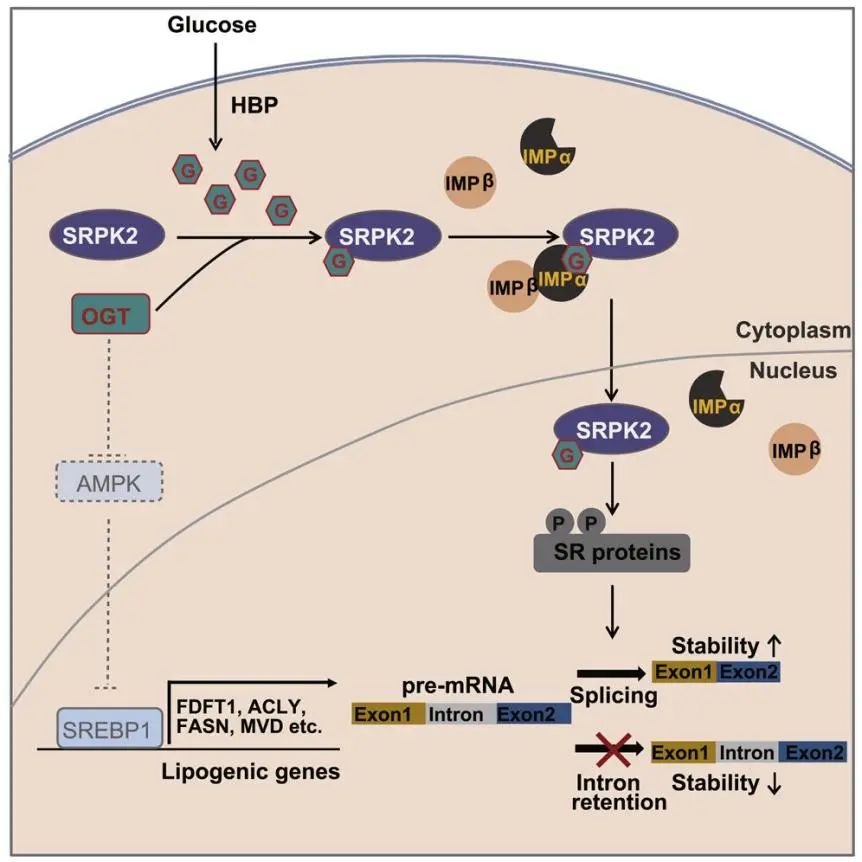
△点击放大图片
本文重点:
1、干扰O-GlcNAc能够阻断脂质从头合成水平
2、O-GlcNAc修饰促进SRPK2在细胞核内的定位
3、importin α蛋白可识别被O-GlcNAc的核定位信号
4、SRPK2的O-GlcNAc修饰促进乳腺癌细胞在体外及体内的生长
总结:脂肪类是细胞生存和生长的必需品。O-GlcNAc修饰通过增强SRPK2的细胞核内定位、在转录后水平上调控肿瘤细胞的脂质从头合成水平,进而促进肿瘤生长;importin α蛋白识别的O-GlcNAc核定位信号在细胞中很常见。
1.HBP在转录后水平上影响脂质从头合成。
HBP是糖酵解的一个分支,负责产生蛋白质O-GlcNAcylation的糖供体UDP-GlcNAc。癌细胞改变了葡萄糖的代谢并导致O-GlcNAcylation的异常, 表明HBP的通量发生了变化。为了研究HBP是否调控脂质从头合成,研究人员通过敲低该途径的关键限速酶GFPT1与回补UDP-GlcNAc的方式,证实了O-GlcNAc修饰可以调控细胞内脂质从头合成水平以及相关基因mRNA的剪接、稳定性与表达水平。
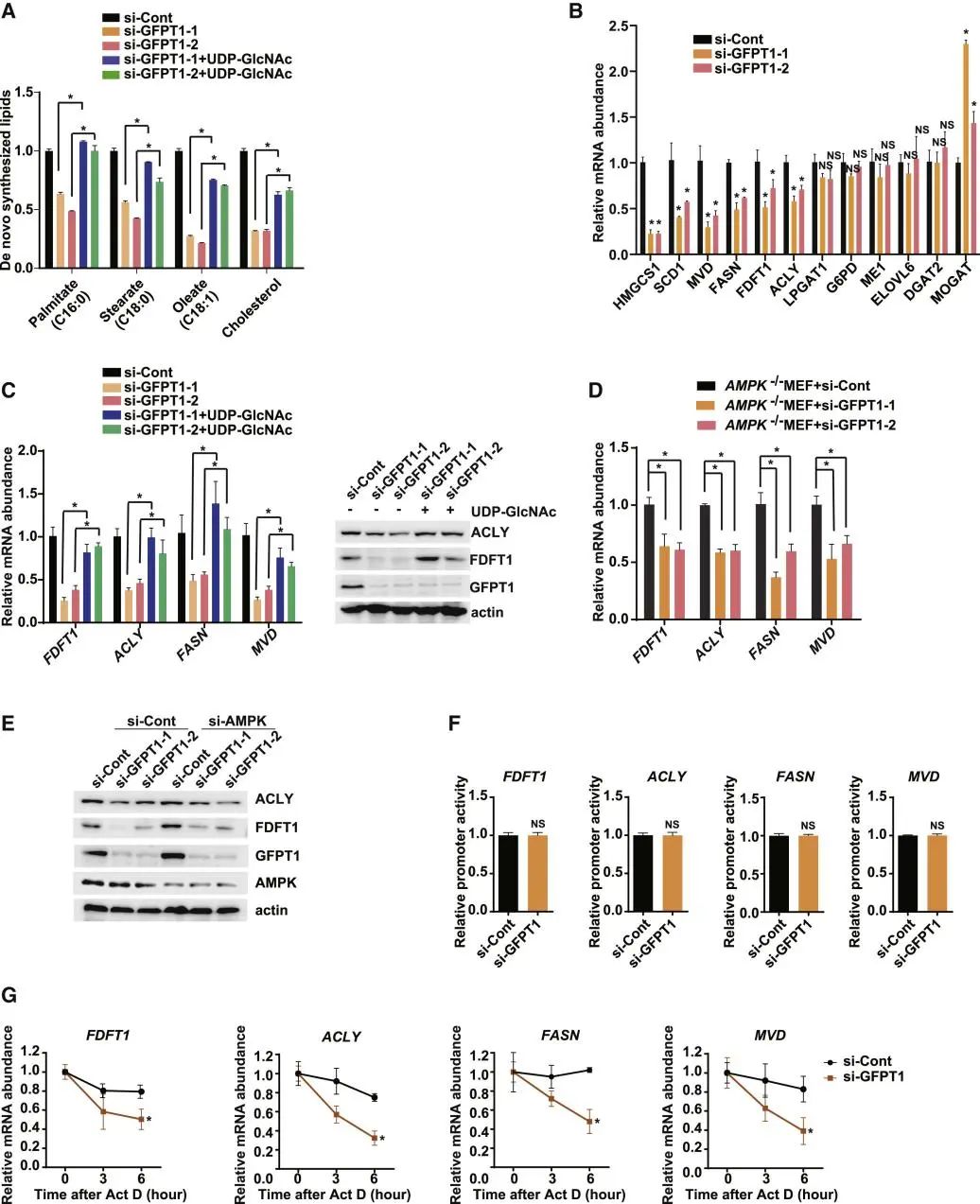
图1: HBP在转录后水平上影响脂质从头合成
△点击放大图片
A. 依据U-13C-葡萄糖的含量,采用液相色谱-质谱(LC-MS)联用法定量分析新的脂质的生成。MCF7细胞被指定的siRNA转染或饥饿处理过夜。根据说明,在隔夜血清饥饿后,将10mM UDP-GlcNAc加入培养基中保持48h。数据以±SEM平均值表示,并表示为相对于转染对照siRNA(si-Cont)细胞数量的倍数变化)。
B. RT-PCR分析MCF7细胞中的脂质合成基因mRNA。将针对GFPT1的SiRNA或si-Cont转染的细胞进行血清饥饿24h。数据以平均±SEM表示,并表示为相对于转染si-Cont的细胞中转录量的倍数变化。*p<0.05(n=3)。
C. MCF7细胞脂质基因的mRNA和蛋白分析。用指定的siRNAs转染细胞,并在血清饥饿过夜后用或不用10mM UDP-GlcNAc处理24h。左:mRNA的丰度测定,并将其表达为相对于转染si-Cont的细胞中转录量的倍数变化。数据以SEM±平均值表示。*p<0.05(n=3)。右:用WB检测ACLY和FDFT1。Actin为内参。
D. AMPK—/—小鼠胚胎成纤维细胞(MEFs)中指定基因的mRNA分析。用指定的siRNA转染细胞,并将血清饥饿处理过夜。转录本采用RT-PCR检测。
E. AMPK缺失 MCF7细胞中的FDFT1和ACLY的WB分析。在血清饥饿处理过夜前,用指定的siRNA转染细胞。Actin为内参。
F. GFPT1被敲除的MCF7细胞中脂质合成基因的启动子活性。在血清饥饿处理过夜前,用启动子结构和指定的siRNA转染MCF7细胞。
G. 定量实时PCR分析GFPT1被敲除的MCF7细胞中脂质合成基因转录本的稳定性。
2.OGT介导SRPK2的O-GlcNAc修饰、核转位及激活。
SRPK2促进了脂质基因mRNA的稳定性和脂质从头合成生成。因此,作者研究了HBP是否通过SRPK2影响新生的脂肪生成。使用CRISPR-Cas9技术生成了SRPK2敲除(KO)细胞,并比较了这些细胞中有无敲除OGT的新的脂质合成发生。
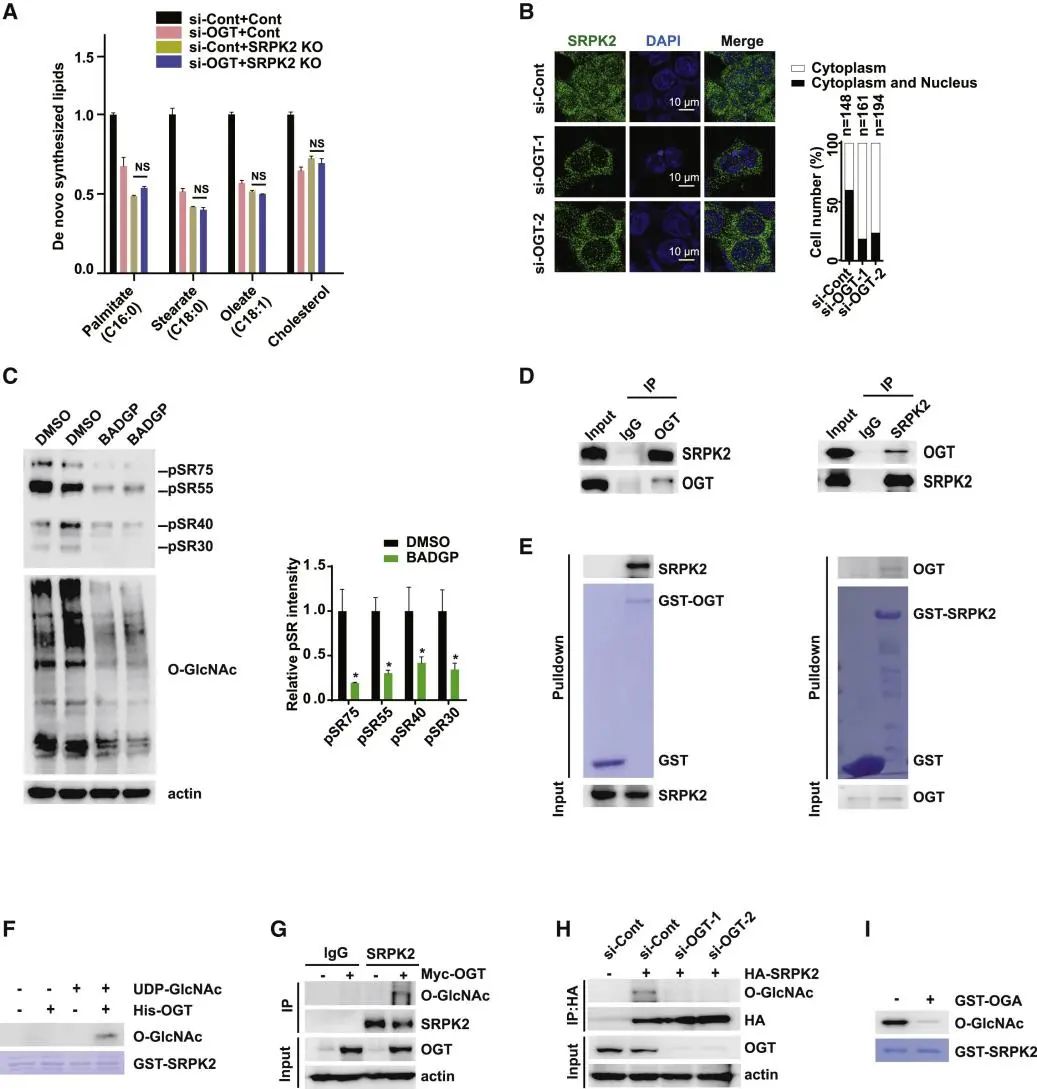
图2:OGT O-GlcNAc修饰 SRPK2并促进其核转位和活化
△点击放大图片
A. LC-MS法定量研究脂质从头合成。对照组或SRPK2敲除(KO)MCF7细胞被指定的siRNA转染。细胞被饥饿处理48小时。图表示脂质从头合成。
B. OGT缺失细胞中SRPK2细胞定位的免疫染色分析。分别用对照组或两种针对OGT的siRNA转染MCF7细胞。左图:代表性图像。右:量化的SRPK2分布。
C. 用BADPG(5mM)或DMSO处理MCF7细胞48h,检测SR蛋白的磷酸化(左)。磷酸化SR蛋白归一化为β-actin的平均带强度的量化(右)。
D. 免疫沉淀(IP)检测HEK293T细胞中OGT与SRPK2的相互作用。
E. Pull-down在体外检测OGT与SRPK2直接相互作用。左:从大肠杆菌(E.coli)细菌中纯化GST- OGT并下拉纯化的SRPK2。右:从大肠杆菌中提取纯化的GST-SRPK2,并下拉纯化的OGT。
F. 以GST-SRPK2(氨基酸[aa]454-521)为底物的体外O-GlcNAcylation试验。
G. 用指定的质粒转染HEK 293T细胞。用anti-SRPK2抗体免疫沉淀细胞裂解物,用指定的抗体测定SRPK2 O-GlcNAcylation。
H. 用指定的siRNA和质粒转染HEK293T细胞,用指定的抗体通过IP和WB检测SRPK2 O-GlcNAcylation。
I. 用OGA在体外去除SRPK2上的O-GlcNAcylation。
3.OGT使SRPK2的S490、T492和T498位点发生是O-GlcNAc修饰,并且促进其核转位。
接下来研究人员利用 LC-MS/MS鉴定到了3个高可信的糖基化修饰位点——S490、T492和T498。免疫荧光实验表明OGT介导的糖基化修饰可以促进SRPK2在细胞核内的定位。
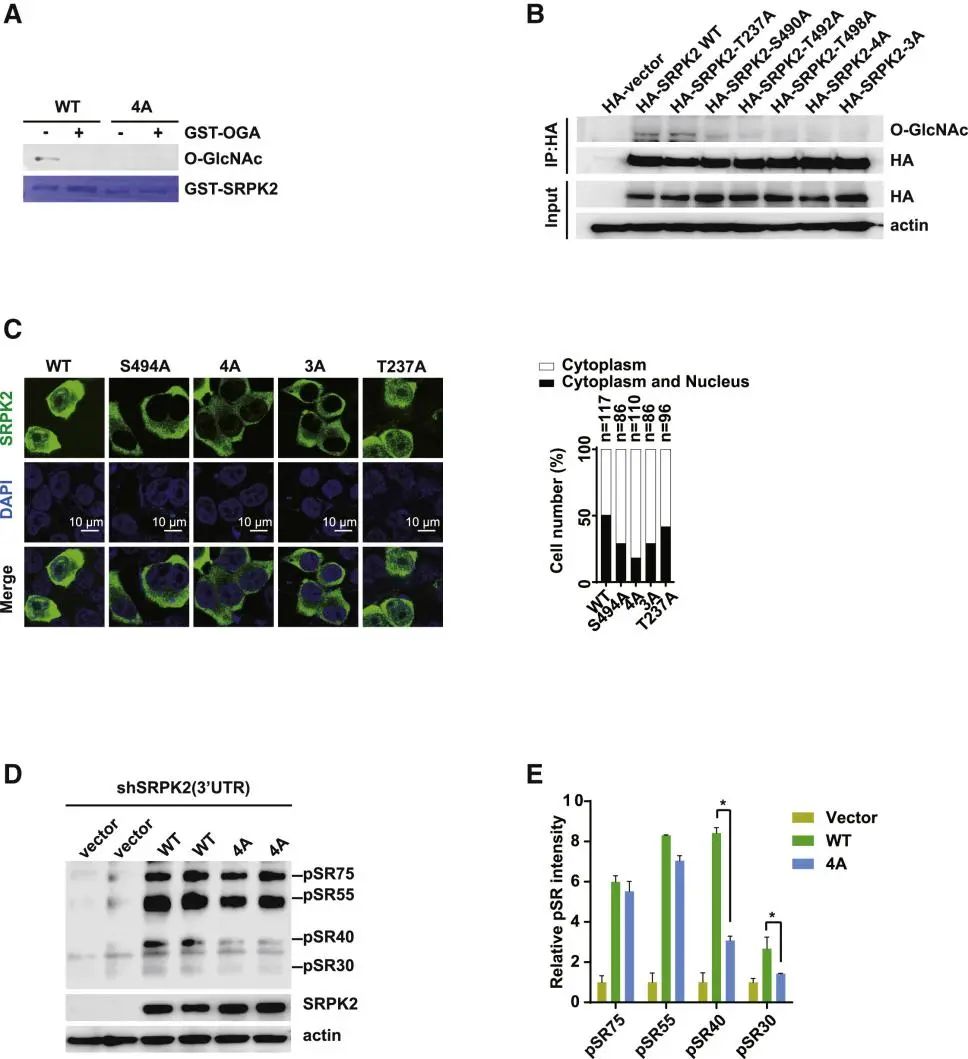
图3:SRPK2 O-GlcNAc修饰诱导其核转位和下游SR蛋白的磷酸化
△点击放大图片
A. 以纯化的GST-SRPK2 WT或4A突变体为底物,用体外O-GlcNA cylation 法鉴定SRPK2 O-GlcNAcylation的修饰位点。
B. 用标记质粒转染HEK 293T细胞,用anti-HA抗体IP分析SRPK2 O-GlcNAcylation, 用标记抗体分析WB。
C. HEK 293T细胞中内源性SRPK2被ShRNA面向的SRPK2 3’ UTR消耗, 然后与cDNA的编码WT 或SRPK2中O-GlcNAcylation缺陷突变体进行稳定的重组(4A或3A或T237A)。用免疫染色法检测SRPK2细胞的定位。
D. HEK 293T细胞中内源性SRPK2被ShRNA面向的SRPK2 3’ UTR消耗, 然后与cDNA的编码WT 或SRPK2中O-GlcNAcylation缺陷突变体进行稳定的重组(4A)。用WB法测定SR蛋白的磷酸化。
E. 定量分析(D)。数据以平均±SEM表示,n=2,*p<0.05。
4.位于核定位序列的SRPK2 O-GlcNAc修饰,能够增强SRPK2 与相应的importin a/b蛋白的相互作用及核转位。
在细胞中,importin α/β系统是调控蛋白入核的经典途径,其中importin α蛋白的N端可以结合importin β蛋白,C端可以结合货物蛋白的核定位序列(Nuclear- localization signal, NLS),从而形成三元复合物通过核孔将蛋白运入细胞核内。所以研究人员通过一系列实验证明,SRPK2上的糖基化修饰位点位于其NLS里面,且体外实验表明糖基化修饰可以显著增强SRPK2与相应importin α蛋白的相互作用。
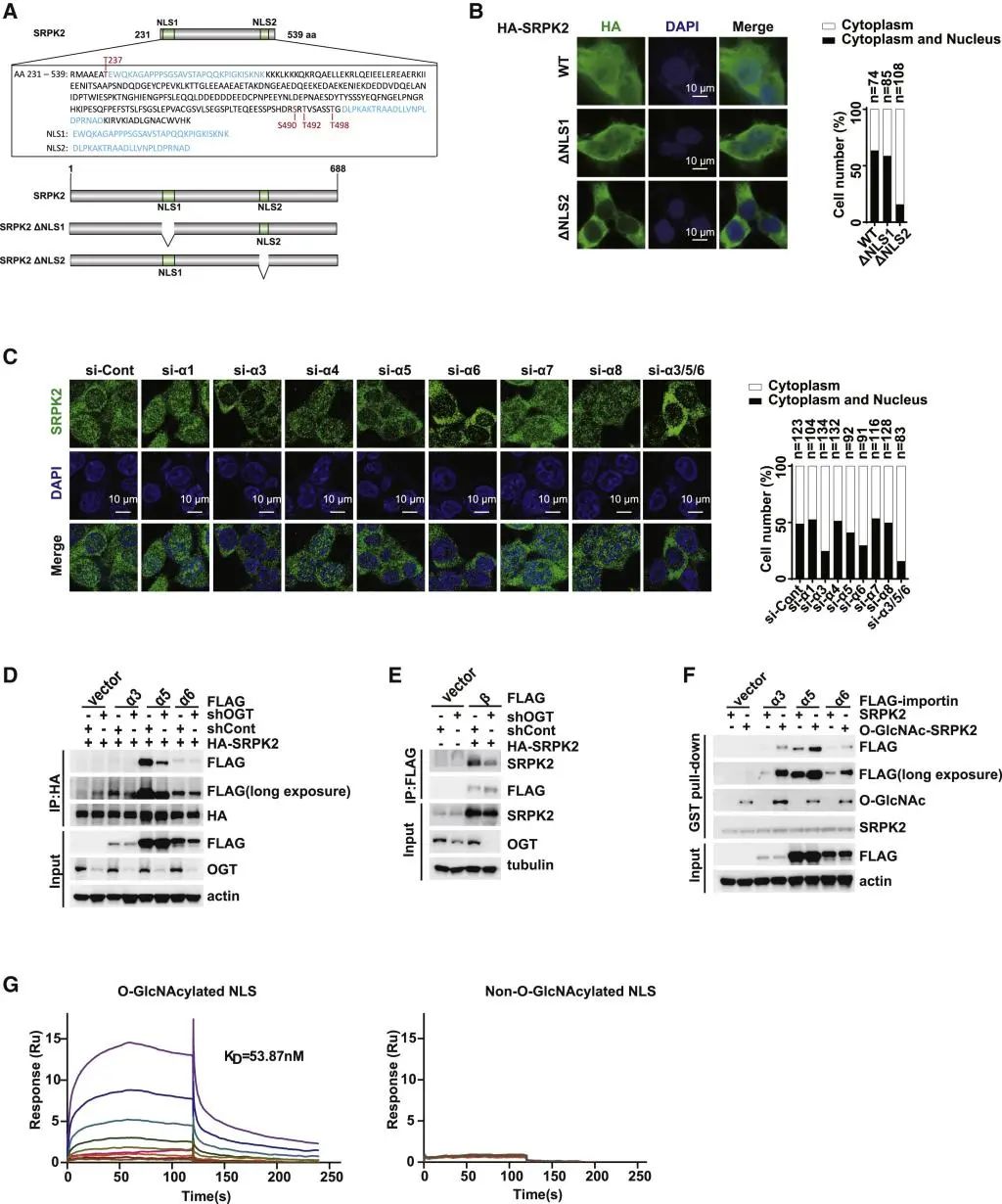
图4::在NLSs上的SRPK2 O-GlcNAcylation诱导其与importin α/β结合
△点击放大图片
A. SRPK2的氨基酸序列公认是核定位序列(NLSs)。公认的NLS1和NLS2的位置以蓝色突出显示,SRPK2 O-GlcNAcylation位点以红色突出显示。NLS的分析源于http://nls-mapper.iab. keio.ac.jp/.
B. MCF7细胞中内源性SRPK2被ShRNA面向的SRPK2 3’UTR消耗,然后被指定的质粒转染,用免疫染色法检测SRPK2细胞的定位。
C. MCF7细胞被指定的siRNA转染,用anti-SRPK2抗体免疫染色,检测SRPK2细胞的定位。
D. 用指定的质粒转染HEK293T细胞,并通过co-IP实验检测importin α3/5/6与SRPK2之间的相互作用。
E. 用指定的质粒转染HEK 293T细胞,并通过co-IP检测importin β与SRPK2的相互作用。
F. 采用体外糖基化测定法生成O-GlcNAcylation的GST-SRPK2(aa 1-540)。将HEK293T细胞转染到指定的importin α或载体中,细胞裂解物由含有或不含O-GlcNAcylation的GST-SRPK2 (aa 1-540)培养。
G. Importin α3与含有或不含O-GlcNAcylation的SRPK2 NLS多肽之间特异性相互作用的SPR结果图。
5.OGT-SRPK2通路平行于mTORC1-S6K1-SRPK2通路。
之后,考虑到前人报道的mTORC1/S6K1介导的SRPK2的磷酸化修饰位点与鉴定到的糖基化位点相近,那么其与糖基化修饰之间是否存在相互影响呢?实验表明,SRPK2糖基化修饰存在与否并不影响SRPK2 S494位点的磷酸化修饰,并且在雷帕霉素处理的条件下,SRPK2糖基化修饰仍然能够响应外界葡萄糖浓度的变化,进一步调控脂质的从头合成水平。这些结果都提示OGT-SRPK2通路平行于mTORC1-S6K1-SRPK2通路。
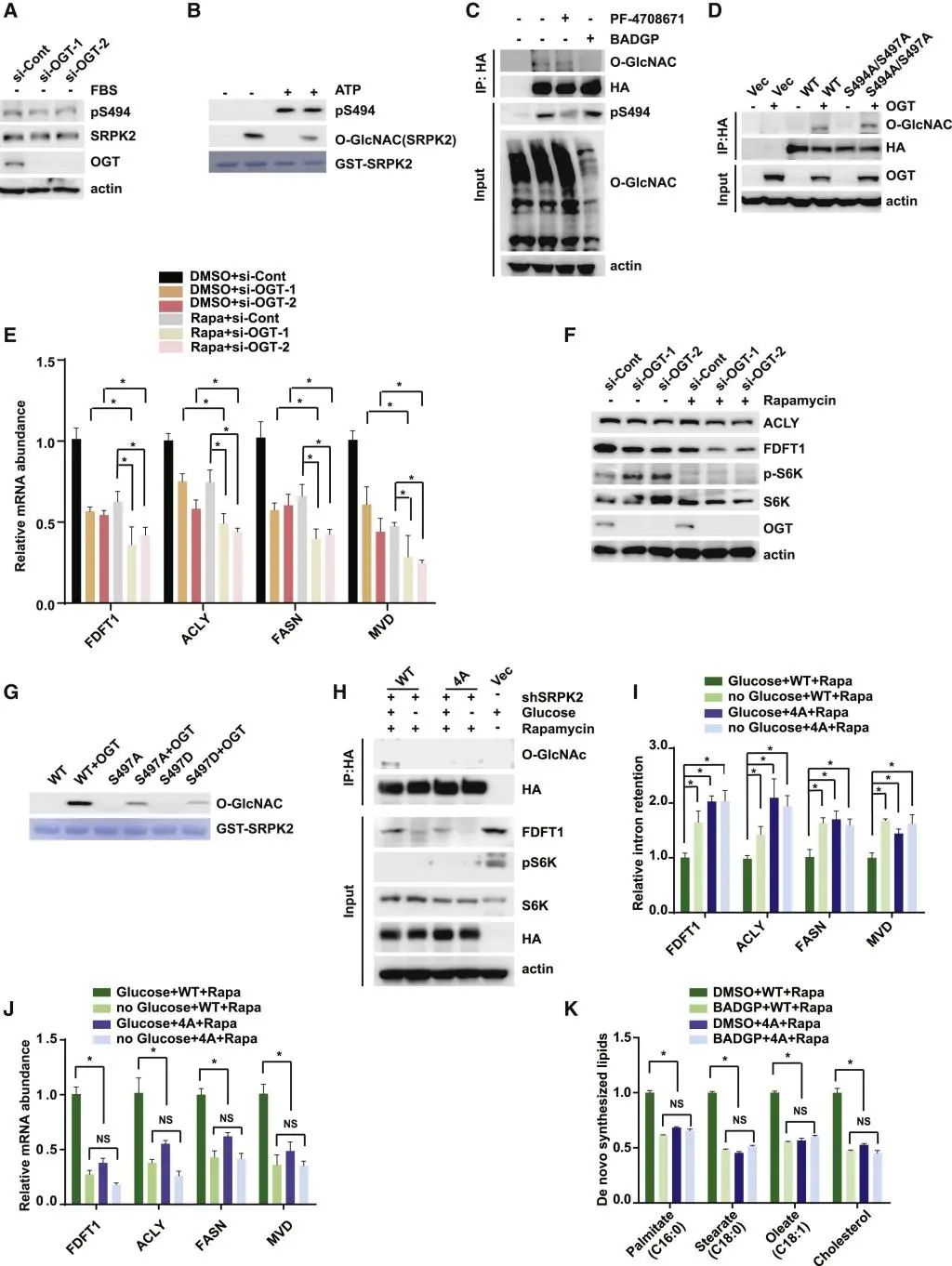
图5:OGT-SRPK2通路平行于mTOR信号通路
△点击放大图片
A. 用指定的siRNA转染HEK293T细胞,然后饥饿处理24h。用WB检测SRPK2的磷酸化。
B. 以未修饰或O-GlcNAcylated SRPK2(aa454-521)为底物的体外S6K1激酶测定。
C. PF-4708671(一种S6K抑制剂,20mM)处理HEK293T细胞24h或BADGP(5mM)处理48h,然后用WB检测SRPK2的O-GlcNAcylation和磷酸化。
D. 用指定的质粒转染HEK293T细胞,并分析SRPK2的O-GlcNAcylation。
E. 用或不用雷帕霉素(100 nM)处理被指定siRNAs转染的MCF7细胞24h,然后用实时qPCR检测指定的mRNA水平。数据以平均±SEM表示;n=3,*p<0.05。
F. 用或不用雷帕霉素(100 nM)处理被指定siRNAs转染的MCF7细胞24h,用WB检测ACLY和FDFT1水平。
G. 以大肠杆菌中纯化的WT或突变型SRPK2(S497A或S497D)蛋白为底物进行体外O-GlcNAcylation试验。
H. 用指定的质粒转染缺失SRPK2的MCF7细胞,在含或不含葡萄糖条件下用雷帕霉素处理48h或不进行处理。用WB检测SRPK2 O-GlcNAcylation、FDFT1和pS6K1水平。
I. 用定量实时qPCR法测定(H)中的相对内含子保留率。数据以平均±SEM表示;n=3,*p<0.05。
J. 通过定量实时PCR测定(H)中相对mRNA的丰度。数据以平均±SEM表示;n=3,*p<0.05。
K. LC-MS法定量研究脂质从头合成。用指定的质粒转染缺失SRPK2的MCF7细胞,用指定的药物处理48h,数据以平均±SEM表示;n=3,*p<0.05。
6.SRPK2的O-GlcNAc修饰促进细胞脂质代谢和肿瘤发生。
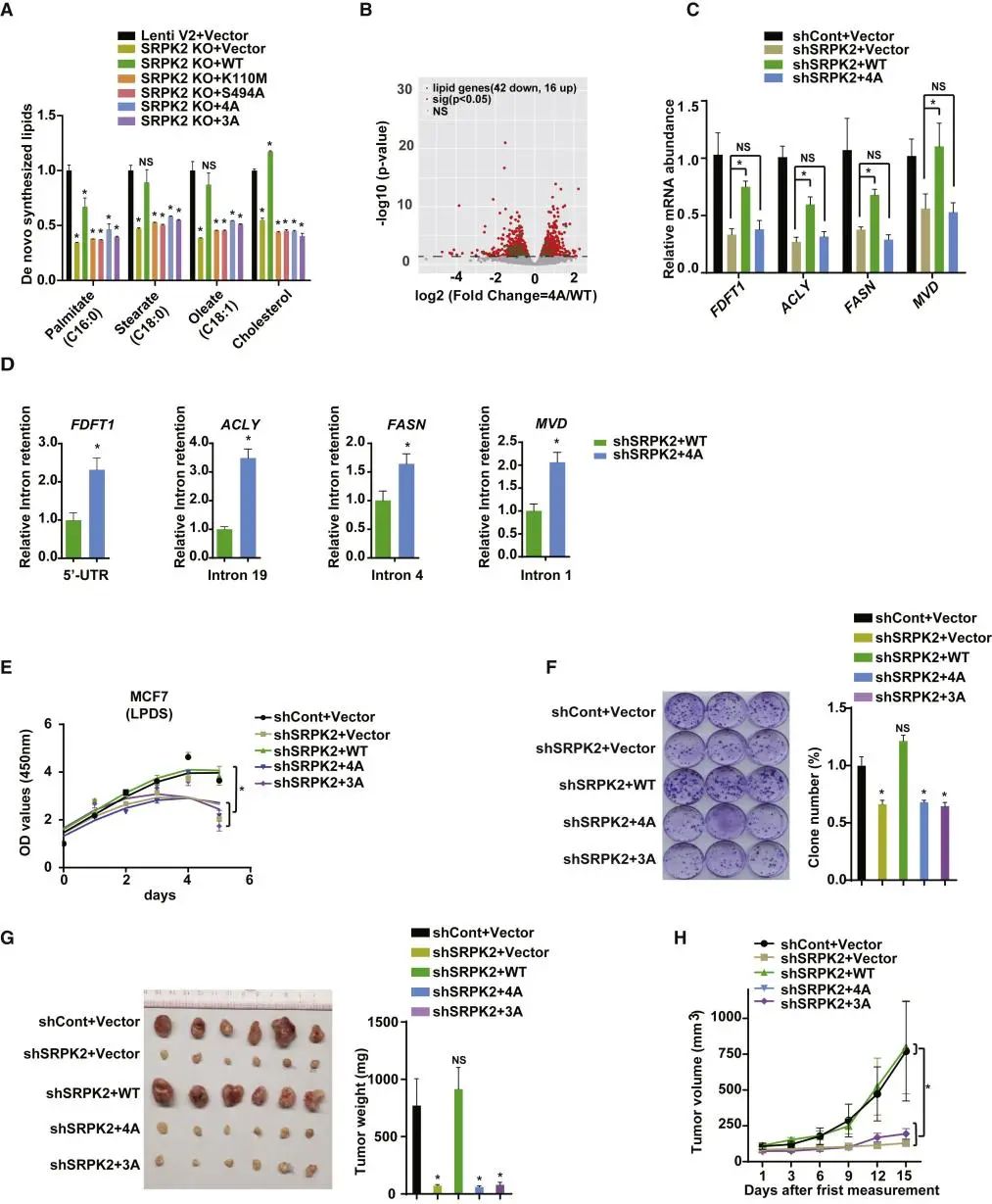
图6:SRPK2 O-GlcNAcylation促进了肿瘤发生
△点击放大图片
A. LC-MS法定量研究脂质从头合成,用指定的质粒转染SRPK2缺失的MCF7细胞,饥饿处理48小时。数据以平均±SEM表示;n=3,*p<0.05。
B. 通过MCF7细胞中的全转录组RNA-seq对WT和4A样品进行分析鉴定脂质基因(绿色) 差异化表达的火山图。用WT SRPK2或4A突变体重组SRPK2缺失的MCF7细胞,然后饥饿处理24h。基因-log10(调整p值)>1.30(与WT组相比)被认为是由SRPK2 O-GlcNAcylation调节的。
C. MCF7细胞内源性SRPK2被ShRNA面向的SRPK2 3’UTR消耗, 然后与cDNA的编码WT 或SRPK2中O-GlcNAcylation缺陷突变体进行稳定的重组。采用RT-PCR法分析脂质基因的mRNA水平。数据以平均±SEM表示;n=3,*p<0.05
D. MCF7细胞指定基因的内含子保留分析。用指定的质粒转染细胞并饥饿处理24小时。
E. 用指定的质粒转染MCF7细胞。用无血清的脂蛋白(LPDS)培养细胞一定天数,用细胞活力分析细胞的增殖。数据以平均±SEM表示;n=3,*p<0.05。
F. 用指定的质粒转染SRPK2缺失的MCF7细胞,并将所得菌落固定,用结晶紫染色。
G. (G和H)OGT介导SRPK2的O-GlcNAcylation促进异种移植小鼠的肿瘤生长。在小鼠死后的终点(G)处称重肿瘤)。图表示第一次测量(H)后的肿瘤体积。
7.由O-GlcNAcylation触发的货物蛋白与importinα结合的核进口蛋白可能是细胞中的一种普遍机制。
最后,研究人员结合自有数据和已发表的数据库信息,发现在三百多个鼠源或者人源蛋白的NLS里面或附近都存在糖基化修饰位点。从这些蛋白中,研究人员选择了RELA与Sp1进行了实验验证,结果证实了这些蛋白的糖基化修饰可以显著增强其与相应importin α蛋白的相互作用,进而促进其在细胞核内的定位。这就说明糖基化修饰促进蛋白入核是细胞内普遍存在的一个重要机制,而importin α蛋白本身很可能是一个O-GlcNAc修饰的“reader”。
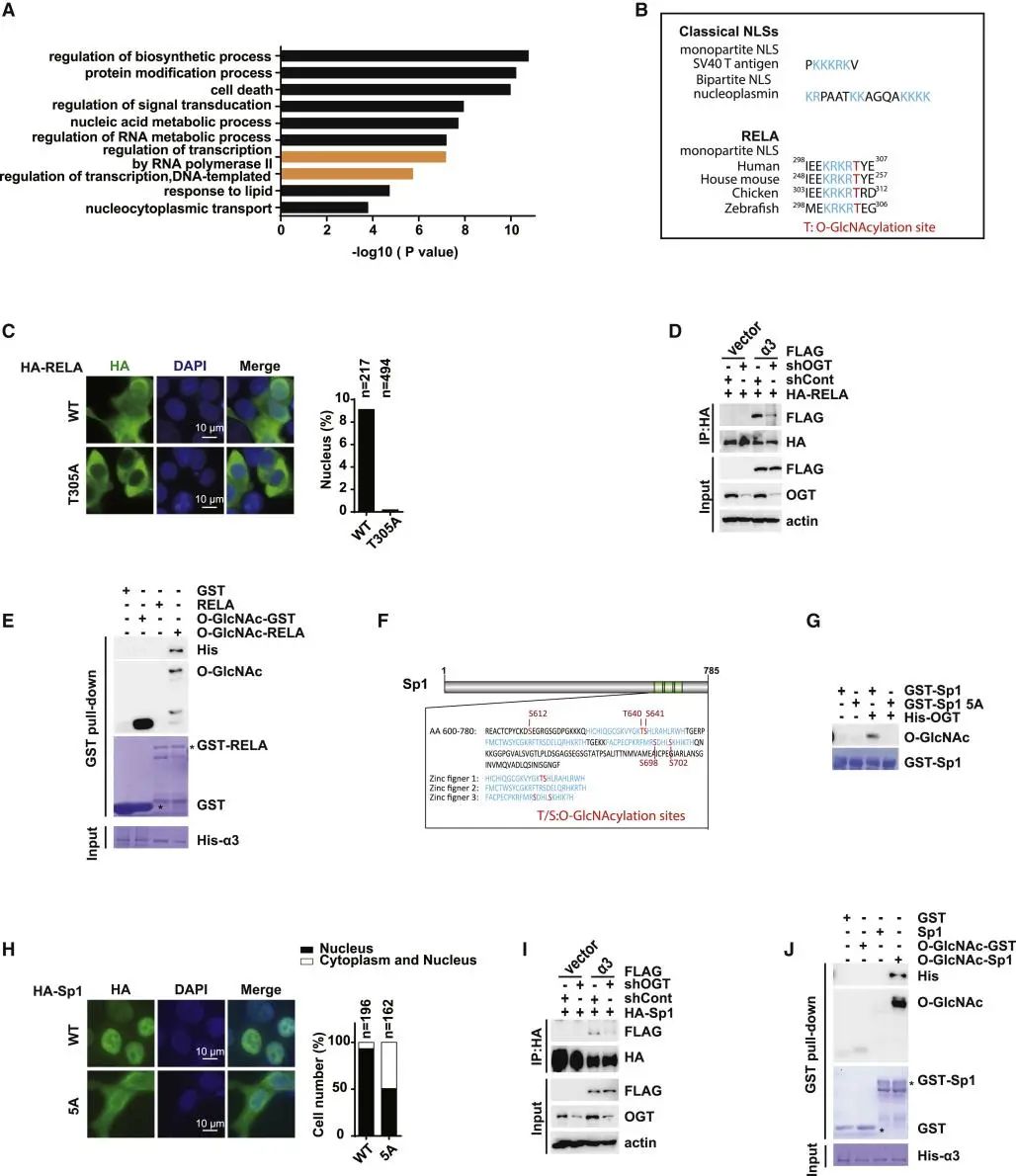
图7:由O-GlcNAcylation触发的货物蛋白与importinα结合的核进口蛋白可能是细胞中的一种普遍机制。
△点击放大图片
A. 小鼠中经O-GlcNAc修饰的NLSs的蛋白基因本体论(GO)富集分析。提出了前10条重要的(p<0.05)富集途径。
B. 几个已知的经典NLSs的氨基酸序列和RELA跨物种的多序列比对。
C. 用指定的质粒转染HEK293T细胞,用免疫染色法检测RELA细胞定位。左图:代表图像。右:左面板的统计分析。
D. 用指定的shRNAs和质粒转染HEK293T细胞,并通过co-IP实验检测importin α3和RELA之间的相互作用。
E. His-importin α从大肠杆菌BL21中纯化得到。并在含有或不含有O-GlcNAcylation的GST-RELA (aa 200-350)中培养。
F. 氨基酸序列的Sp1 3-锌指结构域。
G. 以GST-Sp1(aa 600-780)或GST-Sp1(aa 600-780)5A为底物的体外O-GlcNAcylation试验。
H. 用指定质粒转染HEK293T细胞,用免疫染色法检测Sp1细胞的定位。左图:代表图像。右:左面板的统计分析。
I. 用指定的shRNAs和质粒转染HEK293T细胞,并通过co-IP实验检测importin α3和Sp1之间的相互作用。
J. His-importin α从大肠杆菌BL21中纯化得到。并在含有或不含有O-GlcNAcylation的GST-Sp1(aa 600-780)中培养。。
总的来说,该研究一方面揭示了O-GlcNAc修饰在转录后水平上调控肿瘤细胞脂质从头合成与促进肿瘤生长的分子机制,且OGT-SRPK2通路平行于mTORC1-S6K1- SRPK2通路,提示OGT与mTORC1通路抑制剂的联合使用可能对于相关肿瘤的治疗具有更好的效果;另一方面证实了糖基化修饰介导的importin α蛋白依赖的蛋白入核机制在细胞内普遍存在,提示importin α蛋白很可能是一个O-GlcNAc修饰的“reader”
本文用到的部分产品已为您总结好,请笑纳~
| 货号 | 产品名称 |
| 23708 | Phospho-SRPK2(Ser497)Antibodv |
| 07-1817 | Rabbit polvclonal Anti-pSRPK2(S494) |
| 79409 | SRPK2 Antibody |
| 4332 | ATP-Citrate Lvase Antibodv |
| 2708 | p70 S6 Kinase (49D7)Rabbit mAb |
| 9234 | Phospho-p70 S6 Kinase (Thr389)(108D2)Rabbit mAb |
| 4970 | S-Actin(13E5)Rabbit mAb |
| 2532 | AMPKAntibody |
| 3724 | HA-Tag(C29F4)Rabbit mAb |
| 24083 | oGT (D1D8O)Rabbit mAb |
| 9875 | O-GlcNAc(CTD110.6)Mouse mAb |
| 5322 | GFAT1(D12F4)Rabbit mAb |
| C2527 | BL21(DE3) Competent E.coli. |
| abs810030 | Rapamvcin |
| abs42001607 | PUGNAc |
| abs9232 | BCA蛋白定里试剂盒 |
| abs50003 | CCK-8试剂盒 |
| 208054 | uantNova SYBR Green PCR Kit(500) |
| abs955 | 免疫(共沉淀(PIColP)试剂盒 |
| abs9217 | 苏水素-伊红(HE)染色i试剂盒 |
| abs972 | 胎牛血清(优级 |
| 11995073 | DMEM |
| BR100530 | Series S Sensor Chip CM5.3-pack |
| 17600235 | ProteinAG Sepharose beads |
| 17075605 | Glutathione Sepharose 4B |
| 17524801 | HisTrapHP |
| A-1-3 | GraphPad Prism7 |
02 Nature communacations
PGK1 O-GlcNAc糖基化协调糖酵解和TCA周期,促进肿瘤生长

△点击放大图片
本研究主要揭示了糖酵解途径中的关键酶——磷酸甘油酸激酶1(PGK1)O-GlcNAc糖基化增强糖酵解途径,糖基化修饰通过诱导PGK1的线粒体易位,抑制线粒体内三羧酸(TCA循环),进而促进结肠癌肿瘤的增值和生长。
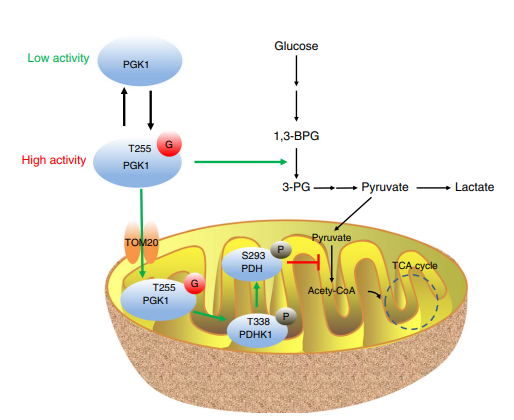
浙江大学生命科学学院易文团队
△点击放大图片
该研究首先研究小鼠结肠癌肿瘤组织中相比于正常组织PGK1的表达明显升高,并且通过敲低各个结肠癌细胞系中的PGK1,反向验证发现糖酵解能力减弱,同时肿瘤细胞的增值和生长也受到抑制。
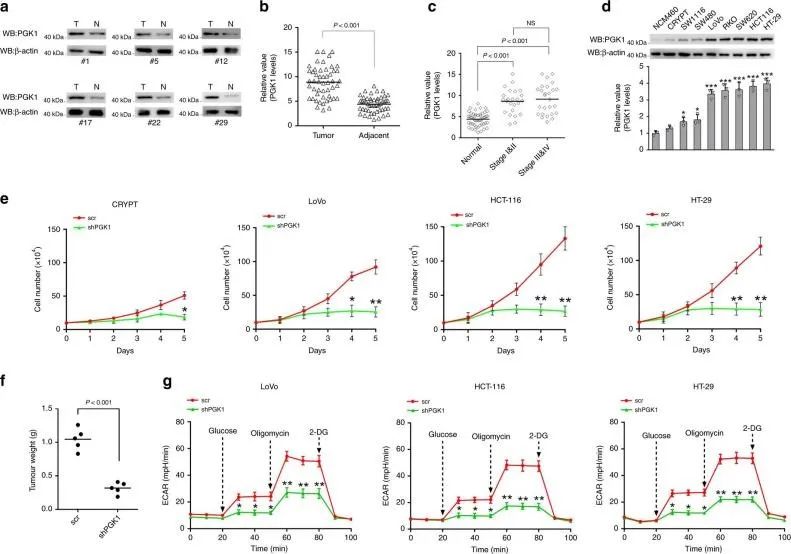
图1:PGK1表达对于结肠癌的发展具有重要意义
△点击放大图片
随后通过生物化学酶标记法、高分辨率质谱分析结合氨基酸定点突变的方法,确定了PGK1具有O-GlcnAc糖基化修饰,而且确定255位的苏氨酸是糖基化修饰的关键位点。
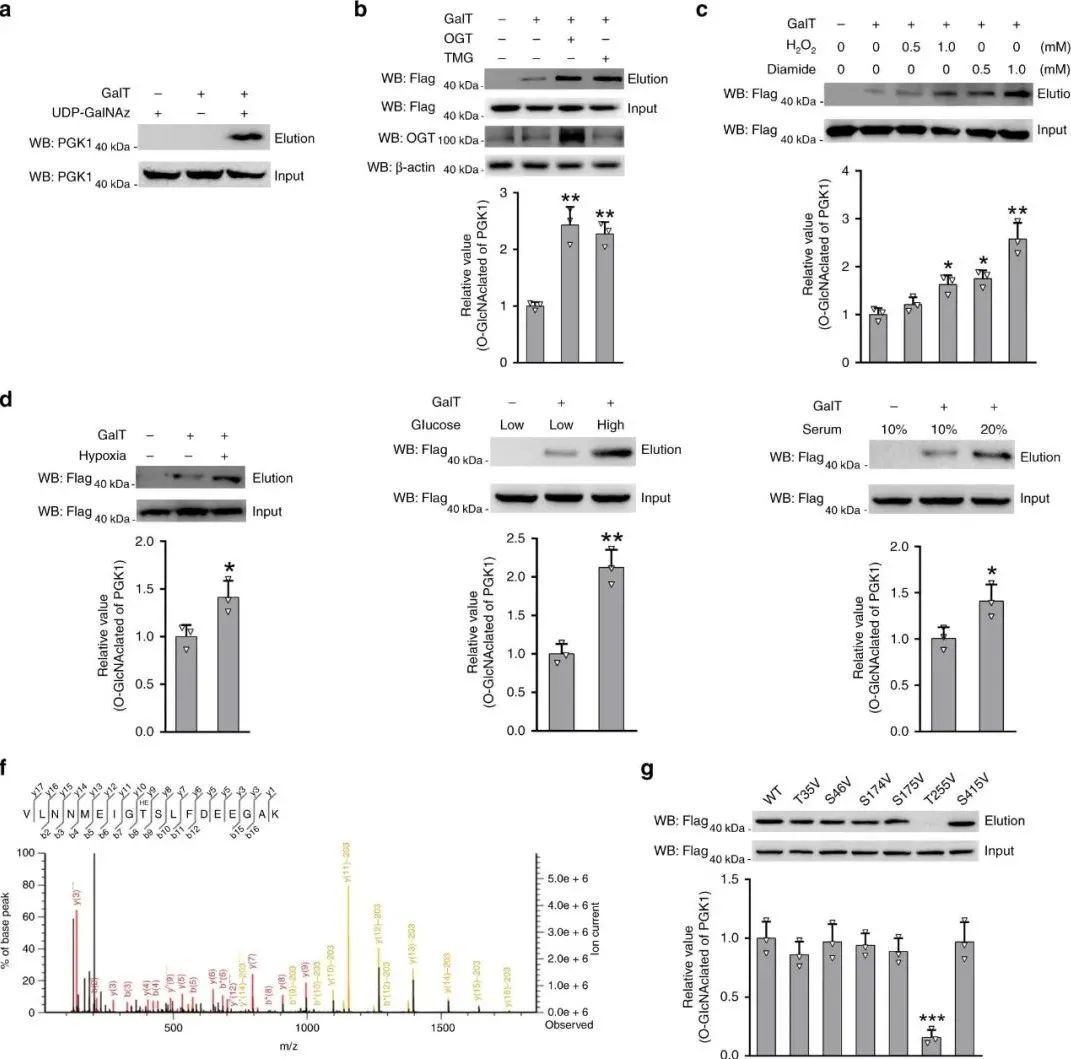
图2 T255是PGK1具有O-GlcnAc糖基化修饰的关键点
△点击放大图片
通过免疫荧光共定位和蛋白质免疫印迹发现,PGK1的O-GlcnAc糖基化修饰可诱导PGK1的线粒体易位,并且对丙酮酸脱氢酶(PDH)磷酸化并使其失活,进而抑制TCA循环。
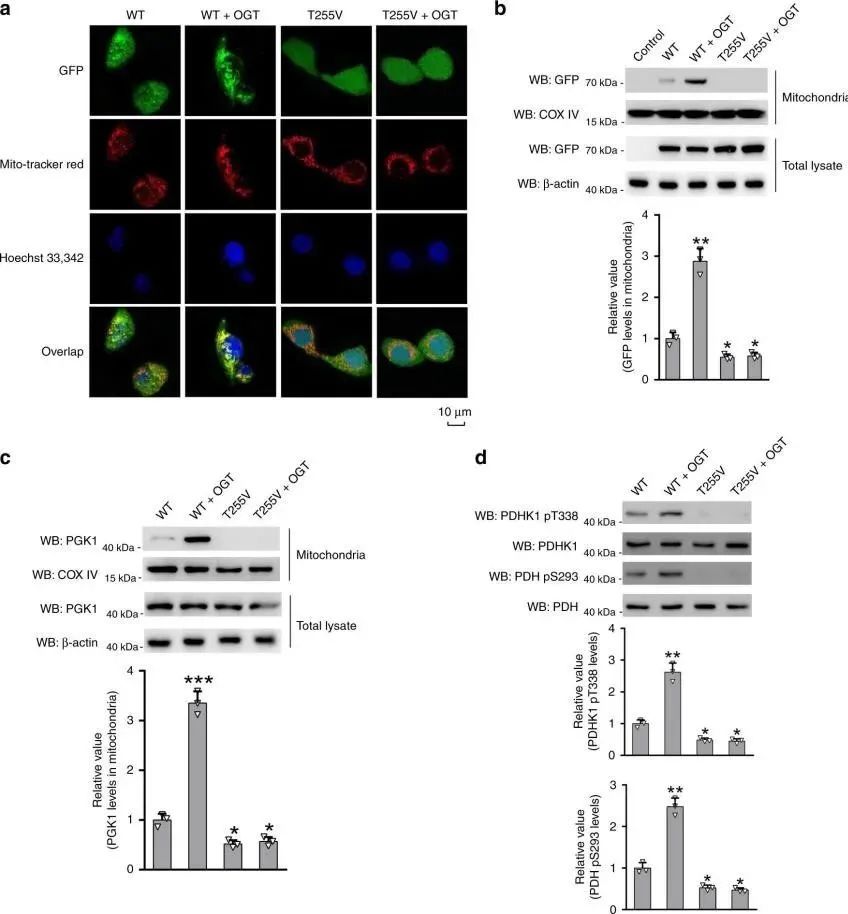
图3 T255 O-GlcNAcylation诱导PGK1的线粒体易位
△点击放大图片
随后对线粒体易位的机制进行了探究,发现T255的糖基化通过促进PGK1和线粒体膜蛋白TOM20的相互作用,促进PGK1的线粒体易位。通过靶向代谢组学分析,发现PGK1糖基化促进糖酵解活性和丝氨酸合成。最后通过小鼠体内成瘤实验临床样本分析,证实PGK1的糖基化修饰可以促进结肠癌的生长。

图4:PGK1的T255 O-GlcNAc化调节细胞内糖代谢
△点击放大图片
本文用到的部分产品已为您总结好,拿走,不谢~~
| 货号 | 产品名称 |
| sC-166432 | anti-PGK1 antibody |
| SAB4200071 | anti-Flag antibody |
| 24083S | anti-OGT antibody |
| 3820S | anti-PDHK1 antibody |
| 42406S | anti-TOM20 antibody |
| 37225 | anti-PIN1 antibody |
| 3205s | anti-PDH antibody |
| orb6670 | anti-PDH(Phospho-Ser293) antibody |
| 11596 | anti-PDHK1(Phospho-Thr338) antibody |
| 4970S | anti-B-actin antibody |
| 4850S | anti-COx IV antibody |
| 2956S | anti-GFP antibody |
| 12698s | anti-His antibody |
| K924 | EZCellTMDirect Glucose Uptake Assay Kit |
| K627 | Lactate Colorimetric Assay Kit lI |
| K678 | Alpha-Ketoglutarate Dehydrogenase Activity Colorimetric Assay Kit |
随着蛋白质糖基化研究越来越多,那目前蛋白质糖基化修饰的检测方法有哪些呢?
1 放射性核素标记法
利用放射性核素标记尿苷二磷酸(UDP)-3H gal是O-GlcNAc糖基化的PTM传统的检测方法,通过在体外与半乳糖苷酶转移酶反应,进行活细胞试验及放射性核素UDP-3H的检测,进而分析蛋白质的糖基化位点。
该法精确度高,但是对样本的需求量大,灵敏度和特异性相对较低,目前通过各种研究也有所改进。
2 植物凝集素法
植物凝集素富集法是利用固定到固相上的凝集素与O-GlcNAc的亲和作用,将O-GlcNAc修饰的蛋白质或肽段富集下来的方法,其中应用最广泛的是麦胚凝集素,麦胚凝集素与含有GlcNAc的糖链具有高度的亲和作用,但是该方法仍然在对于蛋白质的GlcNAc糖基化检测敏感度低的问题。
3 抗体法
利用可以识别 GlcNAc末端和蛋白质O-GlcNAc糖基化修饰抗体,对蛋白质O-GlcNAc糖基化进行识别,该方法优点在于可在不纯化蛋白质的前提下完成对相关蛋白质O-GlcNAc糖基化检测,而且该检测方法具有较高特异性,但是需要纯化多种抗体用于杂交。
4 代谢标记和酶标记富集法
通过利用代谢标记和酶标记的方法将标签引入O-GlcNAc糖基化的肽段上,利用固相中的基团与肽段上的标签结合的亲和特异性,将其从样本中分离出来。例如引入生物素可以用亲和素的固相或抗生物素的抗体进行富集。
5 β-消除-马氏加成法
通过0-糖苷键连接到丝氨酸或苏氨酸上的糖基化修饰,在碱性条件下很容易β-消除,侧链的碳原子和α-碳原子之间形成双键,加入二硫苏糖醇(DTT)后,O-GlcNAc修饰替换成DTT修饰,这种方法叫作β-消除-马氏加成法(BEMAD),该方法,而且后续质谱分析中较稳定,可准确鉴定O-GlcNAc修饰位点。但是该方法丝氨酸和苏氨酸上的其他修饰也容易被β-消除,这样就需要用位点突变等方法做进一步验证。
因此,关于蛋白质糖基化研究方法有很多,对此CST也推出了基于PTMScan®技术的PTMScan® O-GlcNAc糖基化修饰组学研究试剂盒。该试剂盒能够分离、鉴定以及定量大量O-GlcNAc糖基化修饰的细胞肽,具有高特异性和灵敏度,从而可以全面了解细胞和组织样品中的O-GlcNA糖基化修饰,对这些修饰位点发生于何处不会具有偏好性。
产品:PTMScan® O-GlcNAc [GlcNAc-S/T] Motif Kit #95220
PTMScan® O-GlcNAc糖基化修饰组学研究试剂盒采用 Cell Signaling Technology (CST) 的专有方法通过免疫沉淀实现肽富集,这种方法使用一种特殊微珠偶联抗体以及液相色谱法 (LC)-质谱法 (MS/MS) 对细胞蛋白中的O-GlcNAc糖基化修饰位点进行定量分析。

△点击放大图片
用胰蛋白酶消化经 10μM Thiamet-G(TMG)处理 6 小时的HeLa 细胞,并用 PTMScan ® O-GlcNAc [GlcNAc-S/T] 免疫亲和珠进行免疫沉淀分析。Orbitrap Fusion™ Lumos™ 质谱仪分析确定了共 1,235 个不重复位点。