



视频素材来源于YouTube https://www.youtube.com/watch?v=lwReDWWMU0E
版权归原作者所有
大脑是人类最重要的器官之一,其神经中枢系统调控人类各种各样的行为和活动。诸多疾病在大脑的发生发展严重影响人类的健康。
人脑发育的过程存在出几个独特之处,例如神经元复杂性和延伸的增加,已有结果标明这些特征很难在生物模型中进行研究。
因此,模拟人脑发育和疾病进程的体外方法成为科学研究的重点领域。
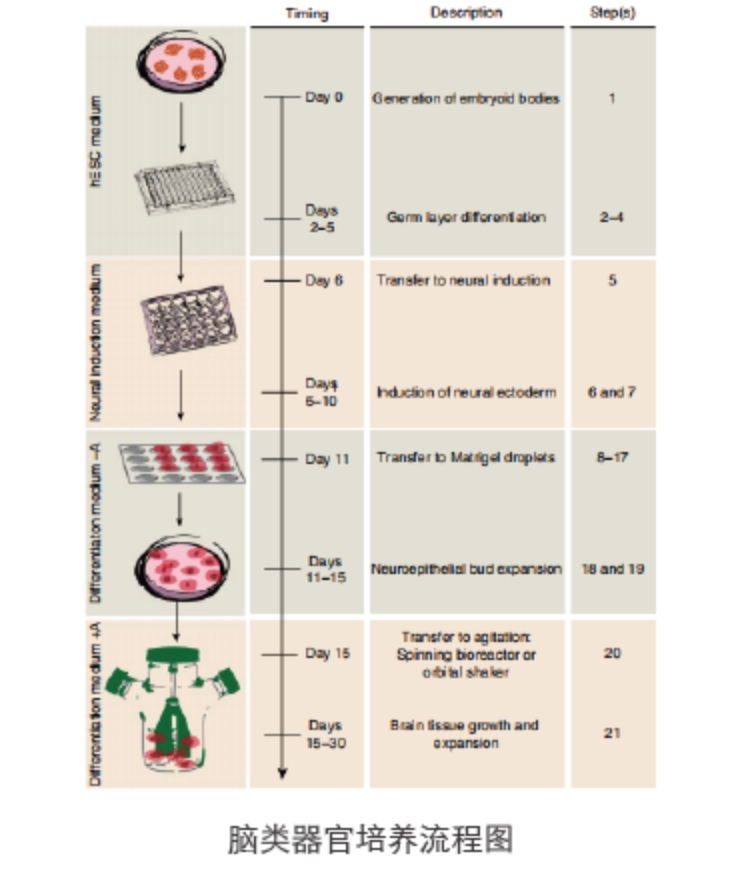
△点击放大图片
实验步骤
- 1
- 2
- 3
- 4
- 5
- 6
- 7
-
培育拟胚体EB
1. 在六孔板的一孔中培育hESC或iPSC克隆,至汇合度为70-80%。一般而言,六孔板的一个孔可以产出大约整个96孔板所需的EB。
【流程A,通过feeder依赖的PSCs培育EB;流程B,通过 feeder非依赖的hESC培育EB。关键步骤:干细胞集落的形态对于脑组织形成的成功至关重要。 菌落应无分化迹象,并应显示多能性的最佳特征。】
流程A:feeder依赖的 ESC/IPSC 培育
(1) 洗涤细胞,去除hESC培养基,加1 ml不含钙和镁的D-PBS。后去除D-PBS,向6孔板的每个孔中加入1 ml酶溶液,然后将细胞放回培养箱中孵育20-30分钟;
(2) 去除分散酶溶液,加入1 mL bFGF的hESC培养基。轻轻敲击培养皿,以去除培养皿上的克隆而不移除MEF和已分化细胞。将含有完整克隆的培养基转移至15 ml锥形管,静置约1分钟使得克隆沉降到试管底部;
(3) 轻轻吸出含有单细胞和MEF的上清液,注意不要干扰贴壁的细胞克隆。另添加1 ml bFGF的hESC培养基,再次使克隆沉降,后去除上清液;
(4) 将克隆重悬于1 ml的胰蛋白酶/ EDTA中,并在37°C下孵育2分钟。加入1 ml的胰蛋白酶抑制剂,并使用1 ml移液器吹打混合溶液, 直到溶液中出现单细胞浑浊为止。取两次5 ul的细胞溶液进行计数,然后加入8 ml bFGF的hESC培养基;
(5) 在室温下以270g离心细胞5分钟,同时加入等体积的台盼蓝标记死细胞。使用血细胞计数器或自动细胞计数器对细胞进行计数。 使用两次计数的平均值;
(6) 首先将细胞重悬于含有ROCK抑制剂(1:100,终浓度50 uM)的1 ml bFGF的hESC培养基中,多次吹打以确保混匀单细胞悬液。然后添加额外适量的带有ROCK抑制剂的 bFGF的hESC培养基,以每150 ul悬液获得9000个活细胞;
(7) 低贴壁型的96孔板,每个孔中加入150 ul悬液,后将其放回培养箱中。
流程B:feeder非依赖的ESC/IPSC 培育
(1) 用1 ml不含钙和镁的D-PBS洗涤细胞,并在六孔板的每个孔添加600 ul不含钙和镁的0.5mM EDTA溶液,后将细胞放回培养箱中孵育4分钟;
(2) 轻轻吸出EDTA溶液而不干扰细胞克隆,后加入1 ml的Accutase酶。将细胞放回培养箱中孵育4分钟;
(3) 采用1 ml的mTeSR1培养基吹打细胞克隆,使其与培养皿分离。取其中2 ml转移到15 ml锥形管中,并使用1 ml移液器吹打混合溶液,直到溶液中出现单细胞浑浊为止。取两次5 ul的细胞溶液进行计数,后添加3 ml mTeSR1培养基并混合均匀;
(4) 在室温下以270g离心细胞5分钟,同时加入等体积的台盼蓝标记死细胞。使用血细胞计数器或自动细胞计数器对细胞进行计数。使用两次计数的平均值;
(5) 首先用1 ml含ROCK抑制剂的bFGF的hESC培养基(1:100,终浓度50 uM)重悬细胞,多次吹打以确保混匀单细胞悬液。然后添加额外适量的带有ROCK抑制剂的bFGF的hESC培养基,以每150 ul悬液获得9000个活细胞;
(6) 低贴壁型的96孔板,每个孔中加入150 ul悬液,后将其放回培养箱中。 -
培育EBs,启动细胞层分化
2. 培育24小时后在组织培养显微镜下观察细胞板,可在镜下观测到边界清晰的小规模EB。继续在组织培养箱中于37°C、5%CO2条件下培养EB。
3. 每隔一天轻轻吸去大约一半体积的培养基,注意不能干扰底部的EB,添加150ul新鲜培养基(最终体积>150ul即可,具体的量并不重要)。加入ROCK抑制剂(1:100)和bFGF培养基“4ng/ml",直到EB开始变亮或直径大于350-400um。可以使用配有照相机和测量软件的倒置显微镜进行尺寸测量。一般而言,在前4d添加ROCK抑制剂和bFGF培养基。
4. 当EB的直径达到350-600um,按照步骤3中所述更换培养基,但不添加ROCK抑制剂和bFGF。 -
诱导原始神经上皮细胞
5. 当EB的直径达到500-600um,并且开始变亮、产生平滑的边缘时(通常是第6天),将每个EB用200ul移液枪头切面切下,移至低附贴壁型的24孔板中,每孔提前加入500ul神经细胞诱导培养基,注意不要破坏EB。
【关键步骤:用无菌剪刀裁剪200ul移液器枪头,以获得直径1‒1.5mm的开口。确保开口不要太小(会破坏EB),但也不要太大(将很难将EB吸到移液器吸头中)。请勿尝试使用刮刀或其他工具将EB从培养板中移出(会破坏EB)。】
6. 将EB转移至24孔板48小时后,再添加500ul神经细胞诱导培养基。
7. 继续培育2天后,在组织培养显微镜上观察EB。此时EB周围应该更亮,表明出现神经外胚层分化。一旦这些区域开始显示与神经上皮分化所一致的假分层上皮放射状的形态(应在神经诱导培养基中培养4-5d后发生),继续进行步骤8,将细胞聚集体转移至基质胶液滴)。健康的细胞聚集体应该具有光滑的边缘。神经上皮在外表面发育,并且在光学上是半透明的。
【关键步骤:当神经上皮出现时,请务必及时进行步骤8,将组织转移至基质胶。不要过早转移组织,否则会影响后续脑组织培养的形态。】 -
将神经上皮组织转移到基质胶液滴中
8. 将基质胶在冰上(4°C)解冻1-2小时。
9. 制作用于制备基质胶液滴的带凹痕封口膜。裁剪一块正方形的封口膜,放置在200ul枪头盒凹痕处,戴手套以摁压封口膜,使其形成一个个小的凹槽。
【注意:因为不能对封口膜进行高压灭菌,因此在准备之前,请确保在清洁的环境中使用封口膜,并用70%(体积/体积)的乙醇对手套和封口膜进行灭菌处理。在此步骤时,培养基中还应包含抗生素以防止污染。】
10. 制作一个包含4×4凹槽的封口膜(总共16个凹槽),并用无菌剪刀将封口膜修剪成一个小正方形。将正方形封口膜放入60mm的组织培养皿中。
【关键步骤:4×4的封口膜大小适合于60mm的组织培养皿。因此,每个60mm培养皿中总包含16个基质胶液滴。】
11. 使用剪切好的200ul移液枪头,将神经上皮组织逐个转移到封口膜的每个凹槽中。
12. 用完整的200ul移液枪头尖端小心吸出液体,以去除每个组织中多余的培养基。
13. 每个组织滴加约30ul的基质胶液滴,充盈整个凹槽,完全浸润组织。
【关键步骤:添加基质胶需迅速,避免组织变干。】
14. 使用10ul移液枪头将每个组织移置于基质胶液滴的中心。
【关键步骤:添加液滴后必须立即进行此步骤,基质胶在室温条件下便会开始凝固。】
15. 将装有基质胶液滴封口膜的60mm培养皿,放回37°C培养箱,孵育20-30分钟以使基质胶聚合。
16. 在60mm培养皿中加入5ml不含维生素A的脑类器官分化培养基。
17. 使用无菌镊子将封口膜翻转过来,基质胶滴浸润在培养基中,再搅动培养基使得所有基质胶滴都从封口膜上脱离下来。继续在二氧化碳培养箱中培养组织液滴。 -
神经上皮芽的固定培养
18. 培育24小时后在显微镜下观察基质胶包埋组织。组织应在1-3d内开始形成更多向外扩张的神经上皮芽,其含有充满液体的空腔。
19. 继续孵育组织24小时,然后用不含维生素A的脑类器官分化培养基培养含有神经上皮组织的基质胶滴,在不搅拌的情况下孵育48小时。【关键步骤:倾斜培养皿,使液滴下沉,并小心吸出培养基,吸取尽可能多的培养基而不干扰基质胶滴,每次采用5ml新鲜培养基替换。】
-
脑组织的生长
20. 静态培养4d后,使用剪切开的1ml移液器吸头(开口约3mm)将培养的类器官转移至125ml的可旋转器官培养瓶中,在75-100ml含有维生素A的脑类器官分化培养基中进行培养。器官培养瓶可以放在培养箱中配套的磁力搅拌板上,也可以放在培养箱中配套的摇床上(振动速度为85r.p.m)。只需将每个60mm培养皿中的培养基替换为含有维生素A的脑类器官分化培养基,然后将培养皿放入培养箱中培养即可。
【关键步骤:不要转移过多的类器官(小于32个),过多的数量会导致类器官融合;确保使用验证应用于组织培养的培养箱低速搅拌板)】
21. 如步骤9所述,全量更换培养基,摇床培养时每3-4天更换,旋转培养瓶每周更换,并实时监测类器官的形态。 -
参考文献
P.S.以上实验步骤均来自文献总结,仅供参考
1.Lancaster, M.A. et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373‒379 (2013). 2.Shevde, N.K. & Mael, A.A. Techniques in embryoid body formation from human pluripotent stem cells. Methods Mol. Biol. 946, 535‒546 (2013).
2.Shevde, N.K. & Mael, A.A. Techniques in embryoid body formation from human pluripotent stem cells. Methods Mol. Biol. 946, 535‒546 (2013).
3.Sasai, Y., Eiraku, M. & Suga, H. In vitro organogenesis in three dimensions: self-organising stem cells. Development 139, 4111‒4121 (2012).
4.Eiraku, M. et al. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 472, 51‒56 (2011).
5.Zhang, S.C., Wernig, M., Duncan, I.D., Brüstle, O. & Thomson, J.A. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 19, 1129‒1133 (2001).
6.Hu, B.-Y. & Zhang, S.-C. Directed differentiation of neural-stem cells and subtype-specific neurons from hESCs. Methods Mol. Biol. 636, 123‒137 (2010).